Electronic properties
What can we learn from calculated MOs?
-
We have seen that we can display a LUMO
-
How reliable are calculated HOMOs and LUMOs?
-
In the HF approach, MOs are filled from the most stable upwards, until
we get to the HOMO, when we have accommodated all the electrons
-
All the rest of the MOs are empty, 'virtual' MOs
-
The most stable empty MO is the LUMO
-
The number of occupied orbitals is fixed by the number of electrons, taken
in pairs
-
If we use a bigger basis set, smaller percentages of more basis functions
will be built in to this fixed number of occupied MOs
-
This should give a better description of each of these MOs, including the
HOMO
-
Total no. of MOs = total no. of basis functions (i.e. contractions
of gaussian primitives)
-
Therefore as we increase the size of the basis set, all the extra MOs are
empty, virtual MOs
-
While the number of occupied MOs depends on the physical reality of how
many electrons there are, the number of unoccupied MOs depends on the model,
not
on physical reality
-
The only requirement on the empty MOs is that they form an orthogonal set
with the filled MOs
-
They are not optimised during the SCF process, because the energy of the
molecule does not depend on them
-
Therefore, unoccupied MOs, including the LUMO, are much less reliable than
occupied MOs, e.g. the HOMO
Natural bond orbitals
-
MOs are delocalised over the whole molecule
-
Usually they bear no resemblance to localised s
and p bonds, or to lone pairs, so they cannot
be used to support familiar chemical reasoning
-
Overlaps between hybrid orbitals to give localised bonds, invented by Pauling
and used by practically all of the chemical world for human explanations
ever since, are (fortunately) not just figments of our collective imagination:
the calculated electron density can also be described in these terms
-
An ordinary MO calculation to get delocalised MOs is done at a high enough
level to reproduce measured geometry or energies, then the atomic basis
set is transformed into an equal number of natural atomic orbitals (NAOs),
and the MOs into an equal number of natural bond orbitals (NBOs)
-
The additional transformations are cheap in computer time
-
This is called NBO analysis
-
It was devised by F. Weinhold, University of Wisconsin, during the 1980s
-
NBO is available in the Gaussian package
-
The process is:
-
AOs
 NAOs
NAOs
-
Basis functions are transformed to natural atomic orbitals
-
NAOs NHOs
-
NAOs are combined into natural hybrid orbitals, so as to describe the atom's
involvement in the calculated molecular electron density
-
The NHOs form orthogonal sets on each atom
-
NHOs NBOs
-
NHOs overlap to give NBOs
-
In this, the total number of orbitals stays the same:
No. of basis functions = no. of MOs = no. of
NAOs = no. of NBOs
Each set consists of normalised linear combinations of the MOs produced
by the original ab initio calculation: each set of orbitals
is an equally valid set of solutions to Schroedinger's equation for the
molecule
-
Because there is not a 1 to 1 correspondence between full or empty MOs
and particular NBOs, the NBOs are not restricted to being either empty
or full, as MOs are
-
NBOs have some fractional occupancy, between 0 and 2
-
Many NBOs contain nearly 2 electrons: these correspond to classical
Lewis-type core or bonding or lone pair orbitals
-
Some of the remaining NBOs are not practically empty: usually these
are antibonding orbitals
-
This corresponds to e.g.
lone pair s*
delocalisation
-
NBO analysis can calculate the stabilisation energy coming from these delocalisations
-
This supports our ideas of donor p bonding,
hyperconjugation, etc.
-
A (crude) contour plotting program, orbplot, is available on the UCS unix
machines for visualising NHOs, NBOs, etc.
-
Although they are produced by objective mathematical transformations of
ab
initio MOs, NBOs look remarkably like the bonding, lone pair, or antibonding
localised orbitals which we teach about
-
One use is to enable visualisation of non-bonded orbital interactions,
e.g.
lone pair - lone pair
which may be critical in determining conformation
-
For an example of some NHOs, NBOs and stabilisation by delocalisation,
see the separate web document NBO plots
-
If you have your unix id set up (see notes in Using
Molden to view a geometry optimisation: it is assumed that you have
done this exercise)
you can look at the corresponding Gaussian .log file which includes
the NBO output
Atomic charges
-
Atomic charge is not a measurable property of a molecule
-
Molecules do not consist of atoms, they consist of nuclei and electrons
-
Some arbitrary decisions have to be taken, on how to divide up calculated
electron density between atoms
-
Consider a MO made of just two AOs, one on each of two atoms A and B
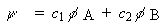
-
The electron density is the square of this:
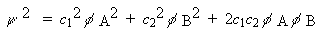
-
To find all the electron(s) in this, we integrate over the whole space
of the molecule:
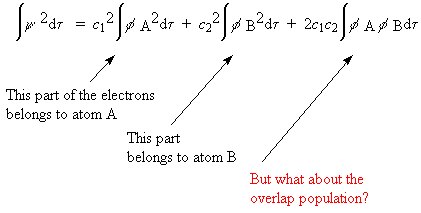
-
In Mulliken population analysis the overlap population is divided
equally between the two partner atoms
-
This is arbitrary!
-
Often, bigger basis sets produce less believable Mulliken charges
-
Most electronic modelling programs calculate charges by the Mulliken method,
because it is simple to do so
-
NBO analysis also calculates atomic charges, by summing occupancy of NAOs,
but the results are different to Mulliken charges
-
A different approach is used to calculate ESP charges
-
Electrostatic potential, produced by the ab initio electron density,
can be calculated at a grid of points
-
All the electron density can be treated equally in this: the only
arbitrary decision is the size of the grid
-
Atomic charges, centred at the nuclei, which would produce the same set
of potentials, are then found by least-squares fitting
-
ESP charges take longer to calculate, but do converge as basis set size
is increased
-
Here are the atomic charges for methyl thiirane, calculated by these three
methods:
Mulliken
NBO ESP
C(H2) -0.521747 -0.55989
-0.342976
C(H) -0.411345 -0.37339
0.038719
S 0.070747
0.11421 -0.192569
C(H3) -0.575628 -0.66843
-0.299563
H 0.222813
0.23979 0.117480
H 0.223591
0.23857 0.094816
H 0.212976
0.23279 0.097235
H 0.254341
0.25257 0.202466
H 0.257897
0.25664 0.170635
H 0.266353
0.26714 0.113758
-
Notice that only the method of fitting the electrostatic potential predicts
a negative sulfur
-
How do we know which method to use?
-
We do not know. The best advice is to report only differences
in charge distribution, between similar compounds
-
These are less likely to depend on method, and may be good enough to explain
or predict trends