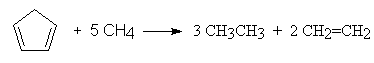- These are called isodesmic reactions
- Examples are:
- proton transfer, e.g.
- redistribution reactions, e.g. transamination or transesterification
- all conformer interconversions
- comparisons between diastereomers
- Isodesmic reaction energies are the easiest to calculate
- The HF method with a medium-sized basis set is sufficient, but a density functional calculation should be better
- DE for the protonation of trimethylamine, above, was calculated as -92 kJ mol-1, at the RHF/6-31G* level, compared with an experimental value of -79 kJ mol-1. A B3LYP/6-31G* calculation reproduced the experimental value. (Hehre, p 145)
Me3N + [NH4]+
Here, there are four N-H bonds, three C-N bonds, and one lone pair, on each side of the equation
- Example:
- hydrogenation, e.g.
- The HF method with a fairly large basis set - at least 6-31G* - is needed
- DF methods with the same size basis set often produce poorer results for these
CH2=CH2 + 2 H2
Here, C-H bonds are formed instead of H-H or C-C bonds, but there are eight bonds altogether on each side of the equation
- Example:
- heterolytic cleavage
- Here, the HF method and a large basis set may work
- Anions have more diffuse electrons, so diffuse functions have to be added to the basis set to allow them to be modelled well
- Beware that free ions are imaginary: the calculation should really be done for complexes of the ions to Lewis acids or bases respectively, e.g. solvent molecules
- This may be too difficult
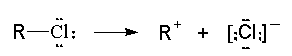
- Example:
- homolytic cleavage
- Here, it is essential to use a method which calculates correlation energy. The Hartree Fock method is not sufficient
ROOR