 © World Health Organization 1999 © World Health Organization 1999
© World Health Organization 1999 © World Health Organization 1999World Health Organization
Department of Noncommunicable Disease Surveillance
Geneva

This document is not a formal publication of the World Health Organization (WHO), and all rights are reserved by the Organization. The document may, however, be freely reviewed, abstracted, reproduced and translated, in part or in whole, but not for sale nor for use in conjunction with commercial purposes.
The views expressed in documents by named authors are solely the responsibility of those authors.
In the late 1970s both WHO (1) and the National Diabetes Data Group (2) produced new diagnostic criteria and a new classification system for diabetes mellitus. This brought order to a chaotic situation in which nomenclature varied and diagnostic criteria showed enormous variations using different oral glucose loads. In 1985 WHO slightly modified their criteria to coincide more closely with the NDDG values (3). There are now many data available, and also much more aetiological information has appeared. It seemed timely to re-examine the issues and to update and refine both the classification and the criteria, and to include a definition of the "Metabolic Syndrome".
An American Diabetes Association (ADA) expert group was convened to discuss these issues. It published its recommendations in 1997 (4). WHO convened a Consultation on the same subject in London, United Kingdom, in December 1996. In general, the ADA and WHO groups reached similar conclusions.
The provisional report of the WHO Consultation (5) solicited comments which were considered in preparing the present report. Both the provisional and the present report were prepared by Professor K.G.M.M. Alberti and Professor P.Z. Zimmet on behalf of the members of the Consultation. The meeting was made possible by generous financial support from Bayer, UK; Bayer, Germany; Novo Nordisk, Copenhagen, Denmark; and The Institute for Diabetes Discovery, New Haven, USA
The term diabetes mellitus describes a metabolic disorder of multiple aetiology characterized by chronic hyperglycaemia with disturbances of carbohydrate, fat and protein metabolism resulting from defects in insulin secretion, insulin action, or both. The effects of diabetes mellitus include long-term damage, dysfunction and failure of various organs. Diabetes mellitus may present with characteristic symptoms such as thirst, polyuria, blurring of vision, and weight loss. In its most severe forms, ketoacidosis or a non-ketotic hyperosmolar state may develop and lead to stupor, coma and, in absence of effective treatment, death. Often symptoms are not severe, or may be absent, and consequently hyperglycaemia sufficient to cause pathological and functional changes may be present for a long time before the diagnosis is made. The long-term effects of diabetes mellitus include progressive development of the specific complications of retinopathy with potential blindness, nephropathy that may lead to renal failure, and/or neuropathy with risk of foot ulcers, amputation, Charcot joints, and features of autonomic dysfunction, including sexual dysfunction. People with diabetes are at increased risk of cardiovascular, peripheral vascular and cerebrovascular disease.
Several pathogenetic processes are involved in the development of diabetes. These include processes which destroy the beta cells of the pancreas with consequent insulin deficiency, and others that result in resistance to insulin action. The abnormalities of carbohydrate, fat and protein metabolism are due to deficient action of insulin on target tissues resulting from insensitivity or lack of insulin.
If a diagnosis of diabetes is made, the clinician must feel confident that the diagnosis is fully established since the consequences for the individual are considerable and lifelong. The requirements for diagnostic confirmation for a person presenting with severe symptoms and gross hyperglycaemia differ from those for the asymptomatic person with blood glucose values found to be just above the diagnostic cut-off value. Severe hyperglycaemia detected under conditions of acute infective, traumatic, circulatory or other stress may be transitory and should not in itself be regarded as diagnostic of diabetes. The diagnosis of diabetes in an asymptomatic subject should never be made on the basis of a single abnormal blood glucose value. For the asymptomatic person, at least one additional plasma/blood glucose test result with a value in the diabetic range is essential, either fasting, from a random (casual) sample, or from the oral glucose tolerance test (OGTT). If such samples fail to confirm the diagnosis of diabetes mellitus, it will usually be advisable to maintain surveillance with periodic re-testing until the diagnostic situation becomes clear. In these circumstances, the clinician should take into consideration such additional factors as ethnicity, family history, age, adiposity, and concomitant disorders, before deciding on a diagnostic or therapeutic course of action. An alternative to blood glucose estimation or the OGTT has long been sought to simplify the diagnosis of diabetes. Glycated haemoglobin, reflecting average glycaemia over a period of weeks, was thought to provide such a test. Although in certain cases it gives equal or almost equal sensitivity and specificity to glucose measurement (6), it is not available in many parts of the world and is not well enough standardized for its use to be recommended at this time.
Diabetes in children usually presents with severe symptoms, very high blood glucose levels, marked glycosuria, and ketonuria. In most children the diagnosis is confirmed without delay by blood glucose measurements, and treatment (including insulin injection) is initiated immediately, often as a life-saving measure. An OGTT is neither necessary nor appropriate for diagnosis in such circumstances. A small proportion of children and adolescents, however, present with less severe symptoms and may require fasting blood glucose measurement and/or an OGTT for diagnosis.
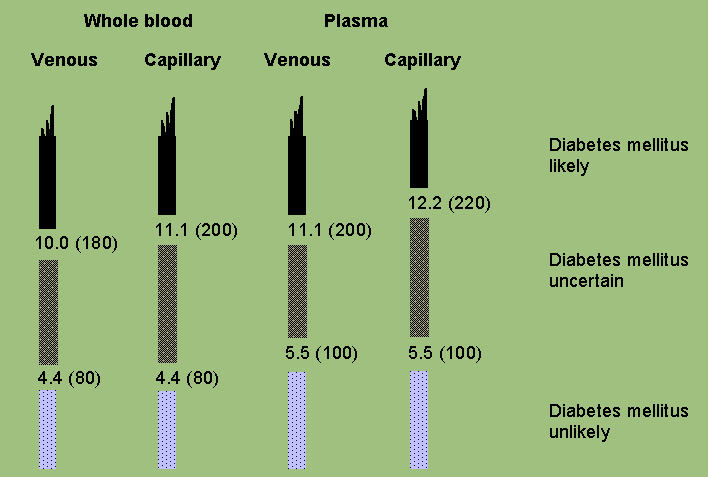
The clinical diagnosis of diabetes is often prompted by symptoms such as increased thirst and urine volume, recurrent infections, unexplained weight loss and, in severe cases, drowsiness and coma; high levels of glycosuria are usually present. A single blood glucose estimation in excess of the diagnostic values indicated in Figure 1 (black zone) establishes the diagnosis in such cases. Figure 1 also defines levels of blood glucose below which a diagnosis of diabetes is unlikely in non-pregnant individuals. These criteria are as in the 1985 report (3). For clinical purposes, an OGTT to establish diagnostic status need only be considered if casual blood glucose values lie in the uncertain range (i.e. between the levels that establish or exclude diabetes) and fasting blood glucose levels are below those which establish the diagnosis of diabetes. If an OGTT is performed, it is sufficient to measure the blood glucose values while fasting and at 2 hours after a 75 g oral glucose load (Annex 1 and Annex 2). For children the oral glucose load is related to body weight: 1.75 g per kg. The diagnostic criteria in children are the same as for adults. Diagnostic interpretations of the fasting and 2-h post-load concentrations in non-pregnant subjects are shown in Table 1.
|
|
Glucose concentration, mmol l-1 (mg dl-1) |
|||
|
|
Whole blood |
Whole blood |
Plasma* |
|
|
|
Venous |
Capillary |
Venous |
|
| Diabetes Mellitus: | ||||
| Fasting | >=6.1 (>=110) | >=6.1 (>=110) | >=7.0 (>=126) | |
|
or |
|
|
|
|
|
2-h post glucose load |
>=10.0 (>=180) |
>=11.1 (>=200) |
>=11.1 (>=200) |
|
|
or both |
||||
|
Impaired Glucose Tolerance (IGT): |
||||
|
Fasting (if measured) |
<6.1 (<110) |
<6.1 (<110) |
<7.0 (<126) |
|
|
and |
||||
|
2-h post glucose load |
>=6.7 (>=120) and |
>=7.8 (>=140) and |
>=7.8 (>=140) and |
|
|
<10.0 (<180) |
<11.1 (<200) |
<11.1 (<200) |
||
|
Impaired Fasting Glycaemia (IFG): |
||||
|
Fasting |
>=5.6 (>=100) and |
>=5.6 (>=100) and |
>=6.1 (>=110) and |
|
|
<6.1 (<110) |
<6.1 (<110) |
<7.0 (<126) |
||
|
and (if measured) |
||||
|
2-h post glucose load |
<6.7 (<120) |
<7.8 (<140) |
<7.8 (<140) |
|
* Corresponding values for capillary plasma are: for Diabetes Mellitus, fasting >=7.0 (>=126), 2-h >=12.2 (>=220); for Impaired Glucose Tolerance, fasting <7.0 (<126) and 2-h >=8.9 (>=160) and <12.2 (<220); and for Impaired Fasting Glycaemia >=6.1 (>=110) and <7.0 (<126) and if measured, 2-h <8.9 (<160).
For epidemiological or population screening purposes, the fasting or 2-h value after 75 g oral glucose may be used alone. For clinical purposes, the diagnosis of diabetes should always be confirmed by repeating the test on another day unless there is unequivocal hyperglycaemia with acute metabolic decompensation or obvious symptoms.
Glucose concentrations should not be determined on serum unless red cells are immediately removed, otherwise glycolysis will result in an unpredictable under-estimation of the true concentrations. It should be stressed that glucose preservatives do not totally prevent glycolysis. If whole blood is used, the sample should be kept at 0-4 °C or centrifuged immediately, or assayed immediately.
The major change recommended in the diagnostic criteria for diabetes mellitus is the lowering of the diagnostic value of the fasting plasma glucose concentration to 7.0 mmol l-1 (126 mg dl-1) and above (3), from the former level of 7.8 mmol l-1 (140 mg dl-1) and above. For whole blood the proposed new level is 6.1 mmol l-1 (110 mg dl-1) and above, from the former 6.7 mmol l-1 (120 mg dl-1).
The new fasting criterion is chosen to represent a value which is at the upper end of the range that corresponds in diagnostic significance in many persons to that of the 2-h post-load concentration, which is not changed. This equivalence has been established from several population-based studies (6-8) and it also represents an optimal cut-off point to separate the components of bimodal frequency distributions of fasting plasma glucose concentrations seen in several populations. Furthermore, several studies have shown increased risk of microvascular disease in persons with fasting plasma glucose concentrations of 7.0 mmol l-1 (126 mg dl-1) and over (6), and of macrovascular disease in persons with such fasting concentrations, even in those with 2-h values of < 7.8 mmol l-1 (140 mg dl-1) (9). Nevertheless, in less obese subjects, in some ethnic groups and in the elderly lower fasting glucose levels may be seen in persons who have 2-h post-load glucose values that are diagnostic for diabetes.
For population studies of glucose intolerance and diabetes, individuals have been classified by their blood glucose concentration measured after an overnight fast and/or 2 h after a 75 g oral glucose load. Since it may be difficult to be sure of the fasting state, and because of the strong correlation between fasting and 2-h values, epidemiological studies or diagnostic screening have in the past been restricted to the 2-h values only (Table 1). Whilst this remains the single best choice, if it is not possible to perform the OGTT (e.g. for logistical or economic reasons), the fasting plasma glucose alone may be used for epidemiological purposes. It has now been clearly shown, however, that some of the individuals identified by the new fasting values differ from those identified by 2-h post glucose challenge values (10,11). The latter include the elderly (12) and those with less obesity, such as many Asian populations. On the other hand, middle-aged more obese patients are more likely to have diagnostic fasting values (10). Overall population prevalence may (13) or may not (7,10,14) be found to differ when estimates using fasting and 2-h values are compared.
The requirements for individual diagnosis differ from those of population studies. The diagnosis should not be based on a single glucose determination but requires confirmatory symptoms or blood/plasma determination. Diagnosis requires the identification of people at risk for development of complications in whom early preventive strategies are indicated. Ideally therefore both the 2-h and the fasting value should be used. These recommendations contrast with those of the ADA Expert Committee which gives primacy to the fasting plasma glucose (4).
The first widely accepted classification of diabetes mellitus was published by WHO in 1980 (1) and, in modified form, in 1985 (3). The 1980 and 1985 classifications of diabetes mellitus and allied categories of glucose intolerance included clinical classes and two statistical risk classes. The 1980 Expert Committee proposed two major classes of diabetes mellitus and named them, IDDM or Type 1, and NIDDM or Type 2. In the 1985 Study Group Report the terms Type 1 and Type 2 were omitted, but the classes IDDM and NIDDM were retained, and a class of Malnutrition-related Diabetes Mellitus (MRDM) was introduced. In both the 1980 and 1985 reports other classes of diabetes included Other Types and Impaired Glucose Tolerance (IGT) as well as Gestational Diabetes Mellitus (GDM). These were reflected in the subsequent International Nomenclature of Diseases (IND) in 1991, and the tenth revision of the International Classification of Diseases (ICD-10) in 1992. The 1985 classification was widely accepted and is used internationally. It represented a compromise between clinical and aetiological classification and allowed classification of individual subjects and patients in a clinically useful manner even when the specific cause or aetiology was unknown. The recommended classification includes both staging of diabetes mellitus based on clinical descriptive criteria and a complementary aetiological classification.
The classification encompasses both clinical stages and aetiological types of diabetes mellitus and other categories of hyperglycaemia, as suggested by Kuzuya and Matsuda (15).
The clinical staging reflects that diabetes, regardless of its aetiology, progresses through several clinical stages during its natural history. Moreover, individual subjects may move from stage to stage in either direction. Persons who have, or who are developing, diabetes mellitus can be categorized by stage according to the clinical characteristics, even in the absence of information concerning the underlying aetiology. The classification by aetiological type results from improved understanding of the causes of diabetes mellitus.
The new classification contains stages which reflect the various degrees of hyperglycaemia in individual subjects with any of the disease processes which may lead to diabetes mellitus.
All subjects with diabetes mellitus can be categorized according to clinical stage, and this is achievable in all circumstances. The stage of glycaemia may change over time depending on the extent of the underlying disease processes (Figure 2). The disease process may be present but may not have progressed far enough to cause hyperglycaemia. The aetiological classification reflects the fact that the defect or process which may lead to diabetes may be identifiable at any stage in the development of diabetes - even at the stage of normoglycaemia. Thus the presence of islet cell antibodies in a normoglycaemic individual makes it likely that that person has the Type 1 autoimmune process. Unfortunately there are few sensitive or highly specific indicators of the Type 2 process at present, although these are likely to be revealed as aetiology is more clearly defined. The same disease processes can cause impaired fasting glycaemia and/or impaired glucose tolerance without fulfilling the criteria for the diagnosis of diabetes mellitus. In some individuals with diabetes, adequate glycaemic control can be achieved with weight reduction, exercise and/or oral agents. These individuals, therefore, do not require insulin and may even revert to IGT or normoglycaemia. Other individuals require insulin for adequate glycaemic control but can survive without it. These individuals, by definition, have some residual insulin secretion. Individuals with extensive beta-cell destruction, and therefore no residual insulin secretion, require insulin for survival. The severity of the metabolic abnormality can either regress (e.g. with weight reduction), progress (e.g. with weight gain), or stay the same.
It is recommended that the terms "insulin-dependent diabetes mellitus" and "non-insulin-dependent diabetes mellitus" and their acronyms "IDDM" and "NIDDM" no longer be used. These terms have been confusing and frequently resulted in patients being classified based on treatment rather than on pathogenesis.
*As additional subtypes are discovered it is anticipated that they will be reclassified within their own specific category.
**Includes the former categories of gestational impaired glucose tolerance and gestational diabetes.
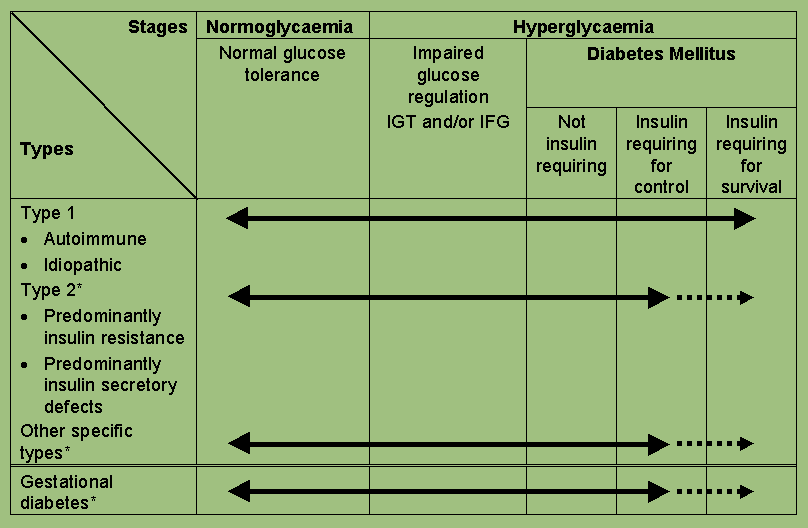
Diabetes mellitus, regardless of underlying cause, is sub-divided into: Insulin requiring for survival (corresponding to the former clinical class of "Insulin Dependent Diabetes Mellitus - IDDM"), e.g. C-peptide deficient; Insulin requiring for control, i.e. metabolic control, rather than for survival, e.g. some endogenous insulin secretion but insufficient to achieve normoglycaemia without added exogenous insulin; and Not insulin requiring, i.e. those who may be controlled satisfactorily by non-pharmacological methods or drugs other than insulin. Together, the latter two sub-divisions constitute the former class of NIDDM.
Impaired glucose regulation (IGT and IFG) refers to a metabolic state intermediate between normal glucose homeostasis and diabetes. It should be stated unequivocally, however, that IFG and IGT are not interchangeable and represent different abnormalities of glucose regulation, one in the fasting state and one post-prandial.
IGT, rather than being a class as in the previous classification, is categorized as a stage in the natural history of disordered carbohydrate metabolism. A stage of IFG is also recognized because such subjects, like those with IGT, have increased risks of progressing to diabetes and macrovascular disease, although prospective data are sparse and early data suggest a lower risk of progression than IGT (18), although a similar CVD risk factor profile has been shown in IFG and IGT subjects (19). IFG refers to fasting glucose concentrations which are lower than those required to diagnose diabetes mellitus but higher than the "normal" reference range.
The values for IFG are a fasting plasma glucose concentration of 6.1 mmol l-1 (110 mg dl-1) or greater (whole blood 5.6 mmol l-1; 100 mg dl-1), but less than 7.0 mmol l-1 (126 mg dl-1) (whole blood 6.1 mmol l-1; 110 mg dl-1). If an OGTT is performed, some individuals with IFG will have IGT or diabetes, but this cannot be determined without an OGTT. If resources allow, it is recommended that all those with IFG have an OGTT to exclude the diagnosis of diabetes.
Individuals who meet criteria for IGT or IFG may be euglycaemic in their daily lives as shown by normal or near-normal glycated haemoglobin levels. IGT and IFG are not clinical entities in their own right, but rather risk categories for future diabetes and/or cardiovascular disease (20,21). They can occur as an intermediate stage in any of the disease processes listed in Table 2. IGT is often associated with the Metabolic Syndrome (Insulin Resistance Syndrome) (22). Thus, IGT may not be directly involved in the pathogenesis of cardiovascular disease, but rather may serve as an indicator or marker of enhanced risk by virtue of its correlation with the other elements of the Metabolic Syndrome that are cardiovascular risk factors. Self-evidently, those individuals with IGT manifest glucose intolerance only when challenged with an oral glucose load.
A fasting venous plasma glucose concentration of less than 6.1 mmol l-1 (110 mg dl-1) has been chosen as "normal" (Table 1). Although this choice is arbitrary, such values are observed in people with proven normal glucose tolerance, although some may have IGT if an OGTT is performed. Values above this are associated with a progressively greater risk of developing micro- and macrovascular complications (8,9,21,23).
The pathological or aetiological processes which often lead to diabetes mellitus begin, and may be recognizable, in some subjects who have normal glucose tolerance. Recognition of the pathological process at an early stage may be useful if progression to more advanced stages can be prevented. Conversely, effective treatments, or occasionally the natural history of some forms of diabetes mellitus, may result in reversion of hyperglycaemia to a state of normoglycaemia. The proposed classification includes a stage of normoglycaemia in which persons who have evidence of the pathological processes which may lead to diabetes mellitus, or in whom a reversal of the hyperglycaemia has occurred, are classified.
The aetiological types designate defects, disorders or processes which often result in diabetes mellitus.
Type 1 indicates the processes of beta-cell destruction that may ultimately lead to diabetes mellitus in which "insulin is required for survival" to prevent the development of ketoacidosis, coma and death. An individual with a Type 1 process may be metabolically normal before the disease is clinically manifest, but the process of beta-cell destruction can be detected. Type 1 is usually characterized by the presence of anti-GAD, islet cell or insulin antibodies which identify the autoimmune processes that lead to beta-cell destruction. In some subjects with this clinical form of diabetes, particularly non-Caucasians, no evidence of an autoimmune disorder is demonstrable and these are classified as "Type 1 idiopathic". Aetiological classification may be possible in some circumstances and not in others. Thus, the aetiological Type 1 process can be identified and sub-categorized if appropriate antibody determinations are performed. It is recognized that such measurements may be available only in certain centres at the present time. If these measurements are performed, then the classification of individual patients should reflect this.
Type 2 is the most common form of diabetes and is characterized by disorders of insulin action and insulin secretion, either of which may be the predominant feature. Both are usually present at the time that this form of diabetes is clinically manifest. By definition, the specific reasons for the development of these abnormalities are not yet known.
Other Specific Types are currently less common causes of diabetes mellitus, but are those in which the underlying defect or disease process can be identified in a relatively specific manner. They include, for example, fibrocalculous pancreatopathy, a form of diabetes which was formerly classified as one type of malnutrition-related diabetes mellitus.
Gestational diabetes is carbohydrate intolerance resulting in hyperglycaemia of variable severity with onset or first recognition during pregnancy. It does not exclude the possibility that the glucose intolerance may antedate pregnancy but has been previously unrecognized. The definition applies irrespective of whether or not insulin is used for treatment or the condition persists after pregnancy.
Women who become pregnant and who are known to have diabetes mellitus which antedates pregnancy do not have gestational diabetes but have "diabetes mellitus and pregnancy" and should be treated accordingly before, during, and after the pregnancy.
In the early part of pregnancy (e.g. first trimester and first half of second trimester) fasting and postprandial glucose concentrations are normally lower than in normal, non-pregnant women. Elevated fasting or postprandial plasma glucose levels at this time in pregnancy may well reflect the presence of diabetes which has antedated pregnancy, but criteria for designating abnormally high glucose concentrations at this time have not yet been established. The occurrence of higher than usual plasma glucose levels at this time in pregnancy mandates careful management and may be an indication for carrying out an OGTT (Annex 1). Nevertheless, normal glucose tolerance in the early part of pregnancy does not itself establish that gestational diabetes may not develop later.
Individuals at high risk for gestational diabetes include older women, those with previous history of glucose intolerance, those with a history of large for gestational age babies, women from certain high-risk ethnic groups, and any pregnant woman who has elevated fasting, or casual, blood glucose levels. It may be appropriate to screen pregnant women belonging to high-risk populations during the first trimester of pregnancy in order to detect previously undiagnosed diabetes mellitus. Formal systematic testing for gestational diabetes is usually done between 24 and 28 weeks of gestation.
To determine if gestational diabetes is present in pregnant women, a standard OGTT should be performed after overnight fasting (8-14 hours) by giving 75 g anhydrous glucose in 250-300 ml water (Annex 1). Plasma glucose is measured fasting and after 2 hours. Pregnant women who meet WHO criteria for diabetes mellitus or IGT are classified as having Gestational Diabetes Mellitus (GDM). After the pregnancy ends, the woman should be re-classified as having either diabetes mellitus, or IGT, or normal glucose tolerance based on the results of a 75 g OGTT six weeks or more after delivery. It should be emphasized that such women, regardless of the 6-week post-pregnancy result, are at increased risk of subsequently developing diabetes. The significance of IFG in pregnancy remains to be established. Any woman with IFG, however, should have a 75 g OGTT.
Patients with any form of diabetes may require insulin treatment at some stage of their disease. Such use of insulin does not, of itself, define the aetiological class.
This form of diabetes, previously encompassed by the terms insulin-dependent diabetes, Type 1 diabetes, or juvenile-onset diabetes, results from autoimmune mediated destruction of the beta cells of the pancreas. The rate of destruction is quite variable, being rapid in some individuals and slow in others (24). The rapidly progressive form is commonly observed in children, but also may occur in adults (25). The slowly progressive form generally occurs in adults and is sometimes referred to as latent autoimmune diabetes in adults (LADA). Some patients, particularly children and adolescents, may present with ketoacidosis as the first manifestation of the disease (26). Others have modest fasting hyperglycaemia that can rapidly change to severe hyperglycaemia and/or ketoacidosis in the presence of infection or other stress. Still others, particularly adults, may retain residual beta-cell function, sufficient to prevent ketoacidosis, for many years (27). Individuals with this form of Type 1 diabetes often become dependent on insulin for survival eventually and are at risk for ketoacidosis (28). At this stage of the disease, there is little or no insulin secretion as manifested by low or undetectable levels of plasma C-peptide (29).
Markers of immune destruction, including islet cell autoantibodies, and/or autoantibodies to insulin, and autoantibodies to glutamic acid decarboxylase (GAD) are present in 85-90 % of individuals with Type 1 diabetes mellitus when fasting diabetic hyperglycaemia is initially detected (30). The peak incidence of this form of Type 1 diabetes occurs in childhood and adolescence, but the onset may occur at any age, ranging from childhood to the ninth decade of life (31). There is a genetic predisposition to autoimmune destruction of beta cells, and it is also related to environmental factors that are still poorly defined. Although patients are usually not obese when they present with this type of diabetes, the presence of obesity is not incompatible with the diagnosis. These patients may also have other autoimmune disorders such as Graves' disease, Hashimoto's thyroiditis, and Addison's disease (32).
There are some forms of Type 1 diabetes which have no known aetiology. Some of these patients have permanent insulinopenia and are prone to ketoacidosis, but have no evidence of autoimmunity (33). This form of diabetes is more common among individuals of African and Asian origin. In another form found in Africans an absolute requirement for insulin replacement therapy in affected patients may come and go, and patients periodically develop ketoacidosis (34).
Diabetes mellitus of this type previously encompassed non-insulin-dependent diabetes, or adult-onset diabetes. It is a term used for individuals who have relative (rather than absolute) insulin deficiency. People with this type of diabetes frequently are resistant to the action of insulin (35,36). At least initially, and often throughout their lifetime, these individuals do not need insulin treatment to survive. This form of diabetes is frequently undiagnosed for many years because the hyperglycaemia is often not severe enough to provoke noticeable symptoms of diabetes (37,38). Nevertheless, such patients are at increased risk of developing macrovascular and microvascular complications (37,38). There are probably several different mechanisms which result in this form of diabetes, and it is likely that the number of people in this category will decrease in the future as identification of specific pathogenetic processes and genetic defects permits better differentiation and a more definitive classification with movement into "Other types". Although the specific aetiologies of this form of diabetes are not known, by definition autoimmune destruction of the pancreas does not occur and patients do not have other known specific causes of diabetes listed in Tables 3-5.
The majority of patients with this form of diabetes are obese, and obesity itself causes or aggravates insulin resistance (39,40). Many of those who are not obese by traditional weight criteria may have an increased percentage of body fat distributed predominantly in the abdominal region (41). Ketoacidosis is infrequent in this type of diabetes; when seen it usually arises in association with the stress of another illness such as infection (42,43). Whereas patients with this form of diabetes may have insulin levels that appear normal or elevated, the high blood glucose levels in these diabetic patients would be expected to result in even higher insulin values had their beta-cell function been normal (44). Thus, insulin secretion is defective and insufficient to compensate for the insulin resistance. On the other hand, some individuals have essentially normal insulin action, but markedly impaired insulin secretion. Insulin sensitivity may be increased by weight reduction, increased physical activity, and/or pharmacological treatment of hyperglycaemia but is not restored to normal (45,46). The risk of developing Type 2 diabetes increases with age, obesity, and lack of physical activity (47,48). It occurs more frequently in women with prior GDM and in individuals with hypertension or dyslipidaemia. Its frequency varies in different racial/ethnic subgroups (47-50). It is often associated with strong familial, likely genetic, predisposition (49-51). However, the genetics of this form of diabetes are complex and not clearly defined.
Some patients who present with a clinical picture consistent with Type 2 diabetes have autoantibodies similar to those found in Type 1 diabetes, and may masquerade as Type 2 diabetes if antibody determinations are not made. Patients who are non-obese or who have relatives with Type 1 diabetes and who are of Northern European origin may be suspected of having late onset Type 1 diabetes.
Several forms of the diabetic state may be associated with monogenic defects in beta-cell function, frequently characterized by onset of mild hyperglycaemia at an early age (generally before age 25 years). They are usually inherited in an autosomal dominant pattern. Patients with these forms of diabetes, formerly referred to as maturity-onset diabetes of the young (MODY), have impaired insulin secretion with minimal or no defect in insulin action (52,53). Abnormalities at three genetic loci on different chromosomes have now been characterized. The most common form is associated with mutations on chromosome 12 in a hepatic nuclear transcription factor referred to as HNF1alpha (54). A second form is associated with mutations in the glucokinase gene on chromosome 7p (55,56). Glucokinase converts glucose to glucose-6-phosphate, the metabolism of which in turn stimulates insulin secretion by the beta cell. Thus, glucokinase serves as the "glucose sensor" for the beta cell. Because of defects in the glucokinase gene, increased levels of glucose are necessary to elicit normal levels of insulin secretion. A third form is associated with a mutation in the HNF4alpha gene on chromosome 20q (57). HNF4alpha is a transcription factor which is involved in the regulation of the expression of HNF1alpha. A fourth variant has recently been ascribed to mutations in another transcription factor gene, IPF-1, which in its homozygous form leads to total pancreatic agenesis (58). Specific genetic defects in other individuals who have a similar clinical presentation are currently being defined.
Point mutations in mitochondrial DNA have been found to be associated with diabetes mellitus and deafness (59). The most common mutation occurs at position 3243 in the tRNA leucine gene, leading to an A to G substitution. An identical lesion occurs in the MELAS syndrome (Mitochondrial myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like syndrome); however, diabetes is not part of this syndrome, suggesting for unknown reasons different phenotypic expressions of this genetic lesion (60).
Genetic abnormalities that result in the inability to convert proinsulin to insulin have been identified in a few families. Such traits are usually inherited in an autosomal dominant pattern (61,62) and the resultant carbohydrate intolerance is mild. Similarly, mutant insulin molecules with impaired receptor binding have been identified in a few families. These are also associated with autosomal inheritance and either normal or only mildly impaired carbohydrate metabolism (63,64).
There are some unusual causes of diabetes which result from genetically determined abnormalities of insulin action. The metabolic abnormalities associated with mutations of the insulin receptor may range from hyperinsulinaemia and modest hyperglycaemia to symptomatic diabetes (65,66). Some individuals with these mutations have acanthosis nigricans. Women may have virilization and have enlarged, cystic ovaries. In the past, this syndrome was termed Type A insulin resistance (65). Leprechaunism and Rabson-Mendenhall syndrome are two paediatric syndromes that have mutations in the insulin receptor gene with subsequent alterations in insulin receptor function and extreme insulin resistance (66). The former has characteristic facial features while the latter is associated with abnormalities of teeth and nails and pineal gland hyperplasia.
Any process that diffusely injures the pancreas can cause diabetes. Acquired processes include pancreatitis, trauma, infection, pancreatic carcinoma, and pancreatectomy (67,68). With the exception of cancer, damage to the pancreas must be extensive for diabetes to occur. However, adenocarcinomas that involve only a small portion of the pancreas have been associated with diabetes. This implies a mechanism other than simple reduction in beta-cell mass (69). If extensive enough, cystic fibrosis and haemochromatosis will also damage beta cells and impair insulin secretion (70,71). Fibrocalculous pancreatopathy may be accompanied by abdominal pain radiating to the back and pancreatic calcification on X-ray and ductal dilatation (72). Pancreatic fibrosis and calcified stones in the exocrine ducts are found at autopsy.
Several hormones (e.g. growth hormone, cortisol, glucagon, epinephrine) antagonize insulin action. Diseases associated with excess secretion of these hormones can cause diabetes (e.g. Acromegaly, Cushing's Syndrome, Glucagonoma and Phaeochromocytoma) (73). These forms of hyperglycaemia typically resolve when the hormone excess is removed.
Somatostatinoma, and aldosteronoma-induced hypokalaemia, can cause diabetes, at least in part by inhibiting insulin secretion (74,75). Hyperglycaemia generally resolves following successful removal of the tumour.
Many drugs can impair insulin secretion. These drugs may not, by themselves, cause diabetes but they may precipitate diabetes in persons with insulin resistance (76,77). In such cases, the classification is ambiguous, as the primacy of beta-cell dysfunction or insulin resistance is unknown. Certain toxins such as Vacor (a rat poison) and pentamidine can permanently destroy pancreatic beta cells (78-80). Fortunately, such drug reactions are rare. There are also many drugs and hormones which can impair insulin action. Examples include nicotinic acid and glucocorticoids (71,72). The list shown in Table 4 is not all-inclusive, but reflects the more commonly recognized drug-, hormone-, or toxin-induced forms of diabetes and hyperglycaemia.
Certain viruses have been associated with beta-cell destruction. Diabetes occurs in some patients with congenital rubella (81). In addition, Coxsackie B, cytomegalovirus and other viruses (e.g. adenovirus and mumps) have been implicated in inducing the disease (82-84).
Diabetes may be associated with several immunological diseases with a pathogenesis or aetiology different from that which leads to the Type 1 diabetes process. Postprandial hyperglycaemia of a severity sufficient to fulfil the criteria for diabetes has been reported in rare individuals who spontaneously develop insulin autoantibodies (85,86). However, these individuals generally present with symptoms of hypoglycaemia rather than hyperglycaemia. The "stiff man syndrome" is an autoimmune disorder of the central nervous system, characterized by stiffness of the axial muscles with painful spasms (87). Affected people usually have high titres of the GAD autoantibodies and approximately one-half will develop diabetes. Patients receiving interferon alpha have been reported to develop diabetes associated with islet cell autoantibodies and, in certain instances, severe insulin deficiency (88).
Anti-insulin receptor antibodies can cause diabetes by binding to the insulin receptor thereby reducing the binding of insulin to target tissues (89). However, these antibodies also can act as an insulin agonist after binding to the receptor and can thereby cause hypoglycaemia (90). Anti-insulin receptor antibodies are occasionally found in patients with systemic lupus erythematosus and other autoimmune diseases (91). As in other states of extreme insulin resistance, patients with anti-insulin receptor antibodies often have acanthosis nigricans. In the past, this syndrome was termed Type B insulin resistance.
Many genetic syndromes are accompanied by an increased incidence of diabetes mellitus. These include the chromosomal abnormalities of Down's syndrome, Klinefelter's syndrome and Turner's syndrome. Wolfram's syndrome is an autosomal recessive disorder characterized by insulin-deficient diabetes and the absence of beta cells at autopsy (92). Additional manifestations include diabetes insipidus, hypogonadism, optic atrophy, and neural deafness. These and other similar disorders are listed in Table 5.
A major classification, diagnostic and therapeutic challenge is the person with hypertension, central (upper body) obesity, and dyslipidaemia, with or without hyperglycaemia. This group of people is at high risk of macrovascular disease (22).
Often a person with abnormal glucose tolerance (IGT or diabetes) will be found to have at least one or more of the other cardiovascular disease (CVD) risk components (22). This clustering has been labelled variously as Syndrome X (22), the Insulin Resistance Syndrome (47), or the Metabolic Syndrome (47).
Epidemiological studies confirm that this syndrome occurs commonly in a wide variety of ethnic groups including Caucasians, Afro-Americans, Mexican-Americans, Asian Indians, Chinese, Australian Aborigines, Polynesians and Micronesians (47,93). In 1988 Reaven focused attention on this cluster, naming it Syndrome X (22). Central obesity was not included in the original description so the term Metabolic Syndrome is now favoured.
Evidence is accumulating that insulin resistance may be the common aetiological factor for the individual components of the Metabolic Syndrome (47,93,94), although there appears to be heterogeneity in the strength of the insulin resistance relationship with different components between, and even within, populations. Alone, each component of the cluster conveys increased CVD risk, but as a combination they become much more powerful (95). This means that the management of persons with hyperglycaemia and other features of the Metabolic Syndrome should focus not only on blood glucose control but also include strategies for reduction of the other CVD risk factors (96).
It is well documented that the features of the Metabolic Syndrome can be present for up to 10 years before detection of the glycaemic disorders (97). This is important in relation to the aetiology of the hyperglycaemia and the associated CVD risk, and the potential to prevent CVD and its morbidity and mortality in persons with glucose intolerance.
The Metabolic Syndrome with normal glucose tolerance identifies the subject as a member of a group at very high risk of future diabetes. Thus, vigorous early management of the syndrome may have a significant impact on the prevention of both diabetes and cardiovascular disease (98).
There is no internationally agreed definition for the Metabolic Syndrome. The following, which does not imply causal relationships, is suggested as a working definition to be improved upon in due course: glucose intolerance, IGT or diabetes mellitus and/or insulin resistance together with two or more of the other components listed below:
Several other components of the Metabolic Syndrome have been described (e.g. hyperuricaemia, coagulation disorders, raised PAI-1, etc.) but they are not necessary for the recognition of the condition.
A clear description of the essential components of the syndrome is needed together with data to support the relative importance of each component. Internationally agreed criteria for central obesity, insulin resistance and hyperinsulinaemia would be of major assistance.
1. WHO Expert Committee on Diabetes Mellitus. Second Report. Geneva: WHO, 1980. Technical Report Series 646.
2. National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. Diabetes 1979; 28: 1039-57.
3. World Health Organization. Diabetes Mellitus: Report of a WHO Study Group. Geneva: WHO, 1985. Technical Report Series 727.
4. The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 1997; 20: 1183-97.
5. Alberti KGMM, Zimmet PZ for the WHO Consultation. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus. Provisional report of a WHO Consultation. Diabetic Medicine 1998; 15: 539-553.
6. McCance DR, Hanson RL, Charles MA, Jacobsson LTH, Pettitt DJ, Bennett PH et al. Comparison of tests for glycated haemoglobin and fasting and two hour plasma glucose concentrations as diagnostic methods for diabetes. BMJ 1994; 308: 1323-28.
7. Finch CF, Zimmet PZ, Alberti KGMM. Determining diabetes prevalence: a rational basis for the use of fasting plasma glucose concentrations? Diabetic Medicine 1990; 7: 603-10.
8. Engelgau MM, Thompson TJ, Herman WH, Boyle JP, Aubert RE, Kenny SJ et al. Comparison of fasting and 2-hour glucose and HbA1c levels for diagnosing diabetes: diagnostic criteria and performance revisited. Diabetes Care 1997; 20: 785-91.
9. Charles MA, Balkau B, Vauzelle-Kervoeden F, Thibult N, Eschwège E. Revision of diagnostic criteria for diabetes (Letter). Lancet 1996; 348: 1657-58.
10. DECODE Study Group on behalf of the European Diabetes Epidemiology Study Group. Will new diagnostic criteria for diabetes mellitus change phenotype of patients with diabetes? Reanalysis of European epidemiological data. BMJ 1998; 317: 371-375.
11. De Vegt F, Dekker JM, Stehouwer CDA, Nijpels G, Bouter LM, Heine RJ. The 1997 American Diabetes Association criteria versus the 1985 World Health Organization criteria for the diagnosis of abnormal glucose tolerance: poor agreement in the Hoorn Study. Diabetes Care 1998; 21: 1686-1690.
12. Wahl PW, Savage PJ, Psaty BM, Orchard TJ, Robbins JA, Tracy RP. Diabetes in older adults: comparison of 1997 American Diabetes Association classification of diabetes mellitus with 1985 WHO classification. Lancet 1998; 352: 1012-1015.
13. Harris MI, Eastman RC, Cowie CC, Flegal KM, Eberhardt MS. Comparison of diabetes diagnostic categories in the US population according to 1997 American Diabetes Association and 1980-1985 World Health Organization diagnostic criteria. Diabetes Care 1997; 20: 1859-62.
14. Ramachandran A, Snehalatha C, Latha E, Vijay V. Evaluation of the use of fasting plasma glucose as a new diagnostic criterion for diabetes in Asian Indian population (Letter). Diabetes Care 1998; 21: 666-67.
15. Kuzuya T, Matsuda A. Classification of diabetes on the basis of etiologies versus degree of insulin deficiency. Diabetes Care 1997; 20: 219-20.
16. Hoet JJ, Tripathy BB, Rao RH, Yajnik CS. Malnutrition and diabetes in the tropics. Diabetes Care 1996; 19: 1014-17.
17. Tripathy BB, Samal KC. Overview and consensus statement on diabetes in tropical areas. Diabetes Metab Rev 1997; 13: 63-76.
18. Shaw JE, Zimmet PZ, de Courten M, Dowse GK, Gareeboo H, Hemraj F et al. IFG or IGT: what best predicts future diabetes? A view of the new ADA recommendations. Diabetes Care 1999 (In press)
19. Larsson H, Berglund G, Lindgärde F, Ahrén B. Comparison of ADA and WHO criteria for diagnosis of diabetes and glucose intolerance. Diabetologia 1998; 41: 1124-1125.
20. Fuller JH, Shipley MJ, Rose G, Jarrett RJ, Keen H. Coronary heart disease risk and impaired glucose tolerance: the Whitehall Study. Lancet 1980; i: 1373-76.
21. Alberti KGMM. The clinical implications of Impaired Glucose Tolerance. Diabet Med 1996; 13: 927-37.
22. Reaven GM. Role of insulin resistance in human disease. Diabetes 1988; 37: 1595-607.
23. McCance DR, Hanson RL, Pettitt DJ, Bennett PH, Hadden DR, Knowler WC. Diagnosing diabetes mellitus - do we need new criteria? Diabetologia 1997; 40: 247-55.
24. Zimmet PZ, Tuomi T, Mackay R, Rowley MJ, Knowles W, Cohen M et al. Latent autoimmune diabetes mellitus in adults (LADA): the role of antibodies to glutamic acid decarboxylase in diagnosis and prediction of insulin dependency. Diabetic Med 1994; 11: 299-303.
25. Humphrey ARG, McCarty DJ, Mackay IR, Rowley MJ, Dwyer T, Zimmet P. Autoantibodies to glutamic acid decarboxylase and phenotypic features associated with early insulin treatment in individuals with adult-onset diabetes mellitus. Diabetic Med 1998; 15: 113-19.
26. Japan and Pittsburgh Childhood Diabetes Research Groups. Coma at onset of young insulin-dependent diabetes in Japan: the result of a nationwide survey. Diabetes 1985; 34: 1241-46.
27. Zimmet PZ. The pathogenesis and prevention of diabetes in adults. Diabetes Care 1995; 18: 1050-64.
28. Willis JA, Scott RS, Brown LJ, Forbes LV, Schmidli RS, Zimmet PZ et al. Islet cell antibodies and antibodies against glutamic acid decarboxylase in newly diagnosed adult-onset diabetes mellitus. Diabetes Res Clin Pract 1996; 33: 89-97.
29. Hother-Nielsen O, Faber O, Sørensen NS, Beck-Nielsen H. Classification of newly diagnosed diabetic patients as insulin-requiring or non-insulin-requiring based on clinical and biochemical variables. Diabetes Care 1988; 11: 531-37.
30. Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Jackson RA et al. Predicting type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 1996; 45: 926-33.
31. Mølbak AG, Christau B, Marner B, Borch-Johnsen K, Nerup J. Incidence of insulin-dependent diabetes mellitus in age groups over 30 years in Denmark. Diabet Med 1994; 11: 650-55.
32. Betterle C, Zanette F, Pedini B, Presotto F, Rapp LB, Monsciotti CM et al. Clinical and subclinical organ-specific autoimmune manifestations in type 1 (insulin-dependent) diabetic patients and their first-degree relatives. Diabetologia 1983; 26: 431-36.
33. McLarty DG, Athaide I, Bottazzo GF, Swai ABM, Alberti KGMM. Islet cell antibodies are not specifically associated with insulin-dependent diabetes in rural Tanzanian Africans. Diabetes Res Clin Pract 1990; 9: 219-24.
34. Ahrén B, Corrigan CB. Intermittent need for insulin in a subgroup of diabetic patients in Tanzania. Diabet Med 1984; 2: 262-64.
35. DeFronzo RA, Bonadonna RC, Ferrannini E. Pathogenesis of NIDDM. In: Alberti KGMM, Zimmet P, DeFronzo RA, eds. International Textbook of Diabetes Mellitus. 2nd edn. Chichester: John Wiley, 1997: pp 635-712.
36. Lillioja S, Mott DM, Spraul M, Ferraro R, Foley JE, Ravussin E et al. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes. Prospective Study of Pima Indians. N Engl J Med 1993; 329: 1988-92.
37. Mooy JM, Grootenhuis PA, de Vries H, Valkenburg HA, Bouter LM, Kostense PJ et al. Prevalence and determinants of glucose intolerance in a Dutch population. The Hoorn Study. Diabetes Care 1995; 18: 1270-73.
38. Harris MI. Undiagnosed NIDDM; clinical and public health issues. Diabetes Care 1993; 16: 642-52.
39. Campbell PJ, Carlson MG. Impact of obesity on insulin action in NIDDM. Diabetes 1993; 42: 405-10.
40. Bogardus C, Lillioja S, Mott DM, Hollenbeck C, Reaven G. Relationship between degree of obesity and in vivo insulin action in man. Am J Physiol 1985; 248: E286-E291.
41. Kissebah AH, Vydelingum N, Murray R, Evans DF, Hartz AJ, Kalkhoff RK et al. Relationship of body fat distribution to metabolic complications of obesity. J Clin Endocrinol Metab 1982; 54: 254-60.
42. Banerji MA, Chaiken RI, Huey H, Tuomi T, Norin AJ, MacKay IR et al. GAD antibody negative NIDDM in adult black subjects with diabetic ketoacidosis and increased frequency of human leukocyte antigen DR3 and DR4: flatbush diabetes. Diabetes 1994; 43: 741-45.
43. Umpierrez GE, Casals MMC, Gebhardt SSP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes 1995; 44: 790-95.
44. Polonsky KS, Sturis J, Bell GI. Non-insulin-dependent diabetes mellitus: a genetically programmed failure of the beta cell to compensate for insulin resistance. N Engl J Med 1996; 334: 777-84.
45. Simonson DC, Ferrannini E, Bevilacqua S, Smith D, Barrett E, Carlson R et al. Mechanism of improvement in glucose metabolism after chronic glyburide therapy. Diabetes 1984; 33: 838-45.
46. Wing RR, Blair EH, Bononi P, Marcus MD, Watanabe R, Bergman RN. Caloric restriction per se is a significant factor in improvements in glycemic control and insulin sensitivity during weight loss in obese NIDDM patients. Diabetes Care 1994; 17: 30-36.
47. Zimmet PZ. Kelly West Lecture 1991: challenges in diabetes epidemiology: from West to the rest. Diabetes Care 1992; 15: 232-52.
48. Harris MI, Cowie CC, Stern MP, Boyko ES, Reiber GE, Bennett PH, eds. Diabetes in America. 2nd edn. Washington DC: US Government Printing Office, 1995 (NIH publ. No. 95-1468).
49. Valle T, Tuomilehto J, Eriksson J. Epidemiology of NIDDM in Europids. In: Alberti KGMM, Zimmet P, DeFronzo RA, eds. International Textbook of Diabetes Mellitus. 2nd edn. Chichester: John Wiley, 1997: pp 125-42.
50. de Courten M, Bennett PH, Tuomilehto J, Zimmet P. Epidemiology of NIDDM in Non-Europids. In: Alberti KGMM, Zimmet P, DeFronzo RA, eds. International Textbook of Diabetes Mellitus. 2nd edn. Chichester: John Wiley, 1997: pp 143-70.
51. Knowler WC, Nelson RG, Saad M, Bennett PH, Pettitt DJ. Determinants of diabetes mellitus in the Pima Indians. Diabetes Care 1993; 16: 216-27.
52. Byrne MM, Sturis J, Menzel S, Yamagata K, Fajans SS, Dronsfield MJ et al. Altered insulin secretory response to glucose in diabetic and nondiabetic subjects with mutations in the diabetes susceptibility gene MODY 3 on chromosome 20. Diabetes 1996; 45: 1503-10.
53. Clement K, Pueyo ME, Vaxillaire M, Rakotoambinina B, Thuillier F, Passa P et al. Assessment of insulin sensitivity in glucokinase-deficient subjects. Diabetologia 1996; 39: 82-90.
54. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY 3). Nature 1996; 384: 455-58.
55. Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO et al. Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes. Nature 1992; 356: 162-64.
56. Vionnet N, Stoffel M, Takeda J, Yasuda K, Bell GI, Zouali H et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes. Nature 1992; 356: 721-22.
57. Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY 1). Nature 1996; 384: 458-60.
58. Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nature Genetics 1997; 117: 138-39.
59. Walker M, Turnbull DM. Mitochondrial related diabetes: a clinical perspective. Diabet Med 1997; 14: 1007-09.
60. Johns DR. Mitochondrial DNA and disease. N Engl J Med 1995; 333: 638-44.
61. Gruppuso PA, Gorden P, Kahn CR, Cornblath M, Zeller WP, Schwartz R. Familial hyperproinsulinemia due to a proposed defect in conversion of proinsulin to insulin. N Engl J Med 1984; 311: 629-34.
62. Robbins DC, Shoelson SE, Rubenstein AH, Tager HS. Familial hyperproinsulinemia: two cohorts secreting indistinguishable type II intermediates of proinsulin conversion. J Clin Invest 1984; 73: 714-19.
63. Haneda M, Polonsky KS, Bergenstal RM, Jaspan JB, Shoelson SE, Blix PM et al. Familial hyperinsulinemia due to a structurally abnormal insulin. Definition of an emerging new clinical syndrome. N Engl J Med 1984; 310: 1288-94.
64. Sanz N, Karam JH, Horita S, Bell GI. Prevalence of insulin-gene mutations in non-insulin-dependent diabetes mellitus. N Engl J Med 1986; 314: 1322-23.
65. Kahn CR, Flier JS, Bar RS, Archer JA, Gorden P, Martin MM et al. The syndromes of insulin resistance and acanthosis nigricans. N Engl J Med 1976; 294: 739-45.
66. Taylor SI. Lilly Lecture: molecular mechanisms of insulin resistance: lessons from patients with mutations in the insulin-receptor gene. Diabetes 1992; 41: 1473-90.
67. Gullo L, Pezzilli R, Morselli-Labate AM, and the Italian Pancreatic Cancer Study Group. Diabetes and the risk of pancreatic cancer. N Engl J Med 1994; 331: 81-84.
68. Larsen S, Hilsted J, Tronier B, Worning H. Metabolic control and B cell function in patients with insulin-dependent diabetes mellitus secondary to chronic pancreatitis. Metabolism 1987; 36: 964-67.
69. Permert J, Larsson J, Westermark GT, Herrington MK, Christmanson L, Pour PM et al. Islet amyloid polypeptide in patients with pancreatic cancer and diabetes. N Engl J Med 1994; 330: 313-18.
70. Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, Adams KS et al. Insulin sensitivity in cystic fibrosis. Diabetes 1994; 43: 1020-26.
71. Phelps G, Chapman I, Hall P, Braund W, Mackinnon M. Prevalence of genetic haemochromatosis among diabetic patients. Lancet 1989; ii: 233-34.
72. Yajnik CS, Shelgikar KM, Naik SS, Kanitkar SV, Orskov H, Alberti KGMM et al. The ketoacidosis-resistance in fibro-calculous-pancreatic-diabetes. Diabetes Res Clin Pract 1992; 15: 149-56.
73. MacFarlane IA. Endocrine diseases and diabetes mellitus. In: Pickup JC, Williams G, eds. Textbook of Diabetes. 2nd edn. Oxford: Blackwell, 1997: pp 64.1-64.20.
74. Krejs GJ, Orci L, Conlon JM, Ravazzola M, Davis GR, Raskin P et al. Somatostatinoma syndrome. N Engl J Med 1979; 301: 285-92.
75. Conn JW. Hypertension, the potassium ion and impaired carbohydrate tolerance. N Engl J Med 1965; 273: 1135-43.
76. Pandit MK, Burke J, Gustafson AB, Minocha A, Peiris AN. Drug-induced disorders of glucose tolerance. Ann Intern Med 1993; 118: 529-40.
77. O'Byrne S, Feely J. Effects of drugs on glucose tolerance in non-insulin-dependent diabetes (parts I and II). Drugs 1990; 40: 203-19.
78. Gallanosa AG, Spyker DA, Curnow RT. Diabetes mellitus associated with autonomic and peripheral neuropathy after Vacor poisoning: a review. Clin Toxicol 1981; 18: 441-49.
79. Esposti MD, Ngo A, Myers MA. Inhibition of mitochondrial complex I may account for IDDM induced by intoxication with rodenticide Vacor. Diabetes 1996; 45: 1531-34.
80. Assan R, Perronne C, Assan D, Chotard L, Mayaud C, Matheron S et al. Pentamidine-induced derangements of glucose homeostasis. Diabetes Care 1995; 18: 47-55.
81. Forrest JA, Menser MA, Burgess JA. High frequency of diabetes mellitus in young patients with congenital rubella. Lancet 1971; ii: 332-34.
82. King ML, Bidwell D, Shaikh A, Voller A, Banatvala JE. Coxsackie-B-virus-specific IgM responses in children with insulin-dependent (juvenile-onset; type 1) diabetes mellitus. Lancet 1983; i: 1397-99.
83. Karjalainen J, Knip M, Hyoty H, Linikki P, Ilonen J, Kaar M-L et al. Relationship between serum insulin antibodies, islet cell antibodies and Coxsackie-B4 and mumps virus-specific antibodies at the clinical manifestation of type 1 (insulin-dependent) diabetes. Diabetologia 1988; 31: 146-52.
84. Pak CY, Eun H, McArthur RG, Yoon J. Association of cytomegalovirus infection with autoimmune type 1 diabetes. Lancet 1988; ii: 1-4.
85. Hirata Y, Ishizu H, Ouchi N et al. Insulin autoimmunity in a case of spontaneous hypoglycaemia. J Jpn Diabet Soc 1970; 13: 312-20.
86. Bodansky HJ, Grant PJ, Dean BM, McNally J, Bottazzo GF, Hambling MH et al. Islet-cell antibodies and insulin autoantibodies in association with common viral infections. Lancet 1986; ii: 1351-53.
87. Solimena M, De Camilli P. Autoimmunity to glutamic acid decarboxylase (GAD) in Stiff-Man syndrome and insulin-dependent diabetes mellitus. Trends Neurosci 1991; 14: 452-57.
88. Fabris P, Betterle C, Floreani A, Greggio NA, de Lazzari F, Naccarato R et al. Development of type 1 diabetes mellitus during interferon alfa therapy for chronic HCV hepatitis (Letter). Lancet 1992; 340: 548.
89. Flier JS. Lilly Lecture: syndromes of insulin resistance: from patient to gene and back again. Diabetes 1992; 41: 1207-19.
90. Kahn CR, Baird KL, Flier JS, Jarrett DB. Effects of autoantibodies to the insulin receptor on isolated adipocytes. J Clin Invest 1977; 60: 1094-106.
91. Tsokos GC, Gorden P, Antonovych T, Wilson CB, Balow JE. Lupus nephritis and other autoimmune features in patients with diabetes mellitus due to autoantibody to insulin receptors. Ann Intern Med 1985; 102: 176-81.
92. Barrett TG, Bundey SE, Macleod AF. Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995; 346: 1458-63.
93. Stern MP. The insulin resistance syndrome. In: Alberti KGMM, Zimmet P, DeFronzo RA, eds. International Textbook of Diabetes Mellitus. 2nd edn. Chichester: John Wiley, 1997: pp 255-83.
94. Haffner SM, Valdez RA, Hazuda HP, Mitchell BD, Morales PA, Stern MP. Prospective analysis of the insulin resistance syndrome (Syndrome X). Diabetes 1992; 41: 715-22.
95. Kaplan NM. The deadly quartet: upper body adiposity, glucose intolerance, hypertriglyceridaemia and hypertension. Arch Intern Med 1989; 149: 1514-20.
96. European Arterial Risk Policy Group on behalf of the International Diabetes Federation (European Region). A strategy for arterial risk assessment and management in Type 2 (non-insulin-dependent) diabetes. Diabet Med 1997; 14: 611-21.
97. Mykkänen L, Kuusisto J, Pyörälä K, Laakso M. Cardiovascular disease risk factors as predictors of Type 2 (non-insulin-dependent) diabetes mellitus in elderly subjects. Diabetologia 1993; 36: 553-59.
98. Eriksson KF, Lindegärde F. Prevention of Type 2 (non-insulin-dependent) diabetes mellitus by diet and physical exercise. Diabetologia 1991; 34: 891-98.
The oral glucose tolerance test (OGTT) is principally used for diagnosis when blood glucose levels are equivocal, during pregnancy, or in epidemiological studies.
The OGTT should be administered in the morning after at least three days of unrestricted diet (greater than 150 g of carbohydrate daily) and usual physical activity. Recent evidence suggests that a reasonable (30-50g) carbohydrate containing meal should be consumed on the evening before the test. The test should be preceded by an overnight fast of 8-14 hours, during which water may be drunk. Smoking is not permitted during the test. The presence of factors that influence interpretation of the results of the test must be recorded (e.g. medications, inactivity, infection, etc.).
After collection of the fasting blood sample, the subject should drink 75 g of anhydrous glucose or 82.5 g of glucose monohydrate (or partial hydrolysates of starch of the equivalent carbohydrate content) in 250-300 ml of water over the course of 5 minutes. For children, the test load should be 1.75 g of glucose per kg body weight up to a total of 75 g of glucose. Timing of the test is from the beginning of the drink. Blood samples must be collected 2 hours after the test load.
Unless the glucose concentration can be determined immediately, the blood sample should be collected in a tube containing sodium fluoride (6 mg per ml whole blood) and immediately centrifuged to separate the plasma; the plasma should be frozen until the glucose concentration can be estimated. For interpretation of results, refer to Table 1.
Reductiometric methods (the Somogyi-Nelson, the ferricyanide and neocuprine autoanalyser methods) are still in use for blood glucose measurement. The o-toluidine method also remains in use but enzyme-based methods are widely available, for both laboratory and near-patient use. Highly accurate and rapid (1-2 min) devices are now available based on immobilized glucose oxidase electrodes. Hexokinase and glucose dehydrogenase methods are used for reference.
Whole blood samples preserved with fluoride show an initial rapid fall in glucose of up to 10 % at room temperature, but subsequent decline is slow; centrifugation prevents the initial fall. Whole blood glucose values are 15 % lower than corresponding plasma values in patients with a normal haematocrit reading, and arterial values are about 7 % higher than corresponding venous values.
The use of reagent-strip glucose oxidase methods has made bedside estimation of blood glucose very popular. However, the cost of the reagent-strips remains high. Some methods still require punctilious technique, accurate timing, and storage of strips in airtight containers. Reasonably quantitative results can be obtained even with visual colour-matching techniques. Electrochemical and reflectance meters can give coefficients of variation of well under 5 %. Reagent-strip methods have been validated under tropical conditions, but are sensitive to extreme climatic conditions. Diabetes may be strongly suspected from the results of reagent-strip glucose estimation, but the diagnosis cannot be confidently excluded by the use of this method. Confirmation of diagnosis requires estimation by laboratory methods.
Patients can easily collect small blood samples themselves (either in specially prepared plastic or glass capillary tubes or on filter-paper), and self-monitoring using glucose reagent-strips with direct colour-matching or meters is now widely practised. Patients should be properly trained in the appropriate techniques to avoid inaccurate or misleading results.
The insulin-treated patient is commonly requested to build up a "glycaemic profile" by self-measurement of blood glucose at specific times of the day (and night). A "7-point profile" is useful, with samples taken before and 90 min after breakfast, before and 90 min after lunch, before and 90 min after an evening meal, and just before going to bed. Occasionally patients may arrange to wake at 0300 h to collect and measure a nocturnal sample. The complete profile rarely needs to be collected within a single 24-hour period, and it may be compiled from samples collected at different times over several days.
Insulin-treated patients who do not have access to facilities for self-measurement of blood glucose should test urine samples passed after rising, before main meals, and before going to bed. Non-insulin-dependent patients do not need to monitor their urine so frequently. Urine tests are of somewhat limited value, however, because of the great variation in urine glucose concentration for given levels of blood glucose. The correlation between blood and urine glucose may be improved a little by collecting short-term fractions (15-30 min) of the urine output. Benedict's quantitative solution or self-boiling, caustic soda/copper sulphate tablets may be used or the more convenient, but costly, semi-quantitative enzyme-based test-strips.
The appearance of persistent ketonuria associated with hyperglycaemia or high levels of glycosuria in the diabetic patient points to an unacceptably severe level of metabolic disturbance and indicates an urgent need for corrective action. The patient should be advised to test for ketone bodies (acetone and aceto-acetic acid) when tests for glucose are repeatedly positive, or when there is substantial disturbance of health, particularly with infections. Rothera's sodium nitroprusside test may be used or, alternatively, reagent-strips that are sensitive to ketones. In emergency situations such as diabetic ketoacidosis, a greatly raised concentration of plasma ketones can be detected with a reagent-strip and roughly quantified by serial 1 in 2 dilution of plasma with water.