Force field methods
-
You should read Chapter 2 of Goodman's 'Chemical Applications of Molecular
Modelling'
about this
-
If two atoms are connected by a spring of equilibrium length rbond
then there will be some potential energy in the spring if it is pulled
out or compressed to some other length r :
Ebond = kbond(r - rbond)2
-
(For the interested, this comes by integrating Hooke's Law, the equation
for a spring balance:
-
For a particular kind of bond, e.g. 4-coord. carbon to 2-coord. oxygen,
we would need to know the stretching constant kbond and
the unperturbed length rbond
-
The method supposes that these are constants over all occurrences of these
elements with these coordination numbers
-
There can be the same kind of energy expression for angle deformation:
Eangle = kangle(q
- qangle)2
-
The constants kangle and qangle
need to be known for the angle at atom B in all possible combinations of
atom types ABC
-
The constants (force field parameters) have been worked out statistically
by fitting models to known structures, and are supplied built in to the
program purchased
-
In force fields in use, more elaborate expressions may be used, to obtain
better reproduction of observed measurements, and there are at least three
more energy terms:
-
EVan der Waals - non-bonded interactions of pairs
of atoms
-
Ebond rotation - correction to account for
rotamer stability beyond what can be explained as Van der Waals repulsion,
e.g. the tendency for 3-coord., p-bonded carbon
to be planar
-
Echarge
- charges are assigned to atoms, based on electronegativities, then energy
of attraction or repulsion of all pairs of atoms is calculated
-
Altogether we have a total force field energy:
Etotal = Ebond + Eangle
+ EVdW + Erotation + Echarge
+ ...??
-
All the energy terms and their parameters form a self-consistent set
-
You can see this if you consider four atoms ABCD:
-
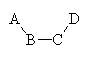 Bond length BC depends
on the Van der Waals interaction of A with D, as well as on the atoms types
of B and C
Bond length BC depends
on the Van der Waals interaction of A with D, as well as on the atoms types
of B and C
-
Therefore the average of all known bond lengths BC is not necessarily the
best value for rbond , because the modeller is going
to use both Ebond and EVdW in
predicting the bond length. If this value of rbond
were used, the predicted bond lengths would come out too long, because
the Van der Waals interaction AD is normally repulsion, to make the BC
bonds longer.
-
The parameters used in the Ebond expression depend on
the parameters used in the EVdW expression, etc.
-
If extra correction terms are added, e.g. for H bonds, to improve the model,
everything has to change
-
The whole set of energy terms and all their parameters, for the whole set
of atom types supported, is called a Force Field
-
Names for some commonly used force fields are:
AMBER, CHARM, COSMIC, MM2, SYBYL, MMFF, DREIDING
-
It is useful to be able to recognise these as force-field methods, in reading
papers and in using modelling programs, where it may not be obvious which
of the provided methods are force-field and which are semi-empirical
-
Because the number of parameters to be supplied with the program increases
rapidly with the number of atom types allowed, force fields are always
restricted to certain elements in certain common environments, where there
are sufficient literature structures to base the parameters on
-
In general, they work well for ordinary organic compounds
-
Some work better for some classes of compound, e.g. strained hydrocarbons,
than others
-
If (in later life) you are about to buy a modelling program, it is best
to find out first whether its supported force fields will work for your
kind of molecule. Manufacturers sometimes supply trial versions,
or trial periods using the real thing, before purchase.
-
It may be possible to add an atom type in some of the programs, but it
is very difficult to get the right values for the parameters, and to do
enough tests to validate the method
-
Many more elements can be dealt with by semi-empirical methods, and all
can be done by ab initio methods, if they have few enough atoms
-
Force field methods are generally not accurate enough to calculate reaction
energies. If it is at all practicable, an electronic method should
be used after the force-field geometry optimisation.
Atom types
-
Different parameters are needed for 3-coord carbon than for 4-coord. carbon,
etc., since the bonds are of different strength and are at different angles
-
Modelling programs try to guess the atom type from its connectivity, but
do not always succeed
-
It is wise to go round a molecule you have constructed, checking that all
the atom types are sensible
-
Modelling programs normally provide some way of finding out atom types
and changing them
-
It may not be obvious to the user what the atom type designation means:
e.g. one program has N sp3, N sp2, N trigonal
as possibilities. Which should you use for N in an organic
amide? Reading the manual may help.