Notes while viewing the model of CH3CHSCH2
(Advice: open two browser windows alongside each other. Go
to this page in one, and the 'Chime model of methyl thiirane' in the other.)
-
I was interested in using this molecule as a source of sulfur atoms because
it will lose sulfur to leave propene
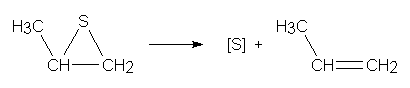
-
I could calculate energies for starting materials and products, for several
possible sulfur acceptors, and successfully predict which would be the
best to try experimentally
-
Unfortunately, there was an alternative mode of reaction: a nucleophilic
iodine atom in my reactant could open the three-membered ring, breaking
one of the C-S bonds. To help assign the product to a particular
isomer, I needed to predict which C would be attacked.
The molecule
(Do the following using the 'Chime model of methyl thiirane')
-
Turn the model to the right and down, to show all atoms
-
Measure the bond angles in the ring, at S and at each C
-
Why is the angle at S so extreme?
The total electron density
(In your other window, go back to the index, and open the 'Chime total
electron density..' model)
-
This takes a little while because a 2 Mb data file of electron density
information has to be downloaded and uncompressed
-
The picture shows a contour of the total electron density
-
In three dimensions, a contour is a surface
-
Make it transparent by
(Right click) Select
Display List
Toggle Transparency
-
In the .spt script file, I chose the value of the electron density for
which the contour was to be plotted, by trial and error
-
Too low a value gives a bland, far-out surface in which detail is lost;
too high a value risks missing out important bits, because there is not
that much density in them
-
Turn the model to look at the sulfur, with the plane of the ring behind
it
-
You can just about imagine the bulges of the lone pairs on sulfur
-
You can clearly see that there is electron density where the bonds are
supposed to be: this may be useful to know in less obvious molecules
-
Other than that, an electron density picture looks like a space-filling
model with VdW radii, which can be got without doing electronic calculations
-
Test this by
(Right mouse click) Display
Spacefill
VdW Radii
-
Return to sticks
(Right mouse click) Display
Sticks
The electrostatic potential
-
Where a nucleophile attacks is partly controlled by where there is a positive
electrostatic potential to attract it
-
One way to display this is to map electrostatic potential energy onto a
total electron density surface, using colours to represent different levels
of potential
(Right click) Select
Display List
Color
Electrostatic Potential
Rasmol
-
These are potentials calculated by Chime, from electronegativities, not
from the electronic structure calculation
-
The white and blue end of the scale is negative potential; the red
end is positive potential
-
Turn the model so that the sulfur is away from you
-
You can see in to a carbon with positive potential, which you can guess
is where a nucleophile will attack
The LUMO
-
A nucleophile must attack an empty MO
-
It will probably attack the Lowest Unoccupied Molecular Orbital
-
In the MO modelling methods, the distribution of individual MOs is also
part of the model
(In your other window, go back to the index, and open the 'Chime LUMO for..'
model)
-
This is the LUMO superimposed on the space-filling model
-
All lobes are colored the same (red) and do not show the sign of the wavefunction.
This is only a limitation of the Chime program: the data is present
in the model.
-
This is clearly a very antibonding orbital because it has several antibonding
nodes
-
Toggle the surface to transparent
-
Find an antibonding nodal surface cutting through the S-C bonds
-
Display sticks
-
Find another node cutting through sulfur and cutting the C-C ring bond
-
Molecular orbitals are delocalised over all or most of the atoms
-
The biggest lobes come from a p orbital on sulfur. We can show that
our nucleophile will not attack there because that is the end of the molecule
with negative potential
-
Map the electrostatic potential, with Rasmol colouring, onto the LUMO surface,
in the same way that you did using the electron density surface
-
Of the warm colour, attractive to a nucleophile, you can see that biggest
accessible LUMO lobe is underneath the ring CH2 carbon
-
This finding guided my further experiments, and was eventually confirmed
by consistent assignment of the carbon and proton NMR spectra of the products