 A first principles study of interstitial Si in diamond
A first principles study of interstitial Si in diamond
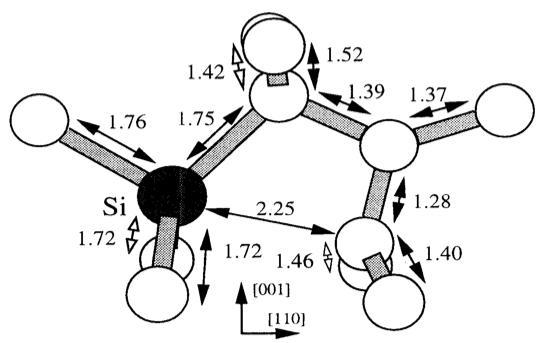
The results of first principles calculations using AIMPRO (a local-density-functional code) are presented to describe the stability of interstitial Si in diamond. It is found that the [100] and [110] split-interstitial configurations are only local minima in the total energy surface. Td interstitial Si reconstructs to form a substitutional Si site close to a self-interstitial. The difference in total energy between the split configuration and the substitutional-Si–self-interstitial complex is more than 6 eV. It is concluded that Si would not adopt an interstitial location in diamond as has been previously suggested from experimental evidence. Interestingly the seif-interstitial remains bound to the substitutional Si with a binding energy around 1 eV.
Go back