 First-principles study of Fe and FeAl defects in SiGe alloys
First-principles study of Fe and FeAl defects in SiGe alloys
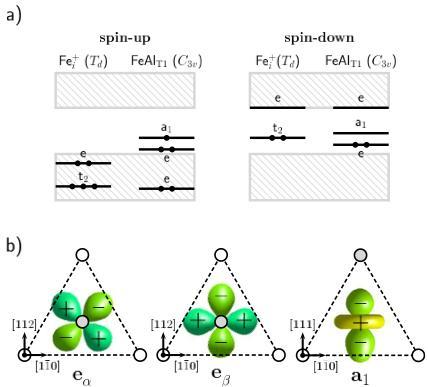
First-principles, spin-polarized local-density-functional calculations are used to model interstitial iron (Fei) and its complexes with substitutional aluminum in dilute SixGe1−x alloys (x<8%). We considered both the effect of direct bonding between Fei or FeiAl with Ge atoms in the x→0 limit and the evolution of the defect properties with the alloy composition. It is found that Fei prefers Si-rich regions, but when placed near a Ge atom, its (0/+) level is shifted toward the conduction band. However, the ionization energy of Fe(+/+2)–Al− is only slightly changed by the presence of neighboring Ge atoms in the proximity. It is also found that indirect alloying effects shift the donor levels of Fei and FeAl at a fast rate toward the valence band. The acceptor levels, however, remain approximately at the same distance from Ev.
Go back