 Charge transfer due to defects in hexagonal boron nitride/graphene heterostructures: An ab initio study
Charge transfer due to defects in hexagonal boron nitride/graphene heterostructures: An ab initio study
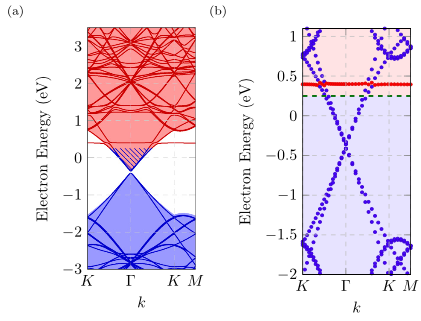
Using density functional theory (DFT), we study charge transfer between hexagonal boron nitride (h-BN) point defects and graphene in h-BN/graphene heterostructures using illustrative examples of intrinsic defects: nitrogen vacancy, boron vacancy, nitrogen antisite, and boron antisite. We show that traditional methods that calculate charge transfer by spatial discrimination of charge to different atoms suffer from the misallocation of charge and introduce an alternative method that relies on the integration of the density of states (DOS). We also show that DFT calculations of charge transfer have cell size dependencies due to a change in the DOS in the vicinity of the defect levels. Our results indicate that the nitrogen and boron anitsites do not participate in charge transfer, whereas the nitrogen and boron vacancies experience the transfer of a whole electron. Additionally, we show that a change in the geometry of a defect corresponds to a change in the charge state of the defect. The results of this paper will be important for a wide variety of device applications that involve charge transfer between h-BN defects and graphene in h-BN/graphene heterostructures, while our methodology can be feasibly extended to a wide range of point defects and heterostructures.
Go back