|
LINKS ICaMB Mol Biol and Bioiformatics resources
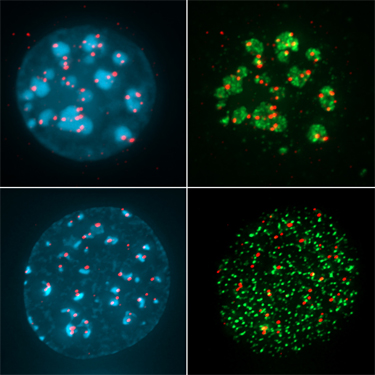
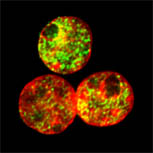
Mouse C127I cell nuclei stained for HP1alpha (green) and CENPB 9red), counterstained with DAPI (blue).
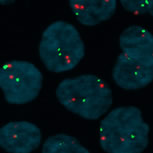
Interphase FISH. MLL locus on chromosome 11(red) and chromosome 11 centromeres (green).
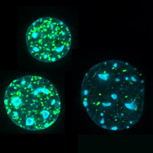
gamma-H2AX foci (green) marking sites of DNA damage induced by etoposide in mouse epithelial cell nuclei. |
Ian
Cowell Institute for Cell and Molecular Biosciences Newcastle University E-mail i.g.cowell@ncl.ac.uk
Research
Interests - The genetic material of eukaryotic cells is organized into chromosomes. These highly organized but dynamic structures support the stable maintenance and faithful replication of the genome, while at the same time permitting transcription and the execution of genetic programs. How these processes occur and how they are coordinated are fundamental questions in modern biology, with great significance for the understanding of conditions such as cancer and aging. Mechanisms of chromosomal translocation in leukaemia. Therapy-related leukaemia is a rare but unfortunate side effect of otherwise successful treatment for primary cancers using DNA damage-inducing anti-cancer drugs. Leukaemia cells from these cases display chromosome aberrations, including characteristic recurrent chromosome translocations, and these genetic lesions are thought to be causative or at least early contributory events in the generation of these leukaemias. How and why recurrent translocations occur in therapy-related and de novo leukaemia is unknown, but the mechanism presumably involves chromosome breakage and incorrect rejoining. To better understand this process we are using interphase in situ hybridization (iFISH) and immunological techniques to probe the chromosome and chromatin dynamics of the regions involved. This work is carried out in Prof. Caroline Austin's laboratory as part of a Leukaemia and Lymphoma Research program grant. Biology of topoisomerase II-mediated DNA damage. Topoisomerase II is an essential enzyme that allows the passage of one DNA duplex through another. This involves an enzyme-bridged DNA double-strand break (DSB) which is normally transient and which is re-ligated after the passage of the second duplex. This enzyme-bridged break is stabilized by topoisomerase II poisons, and the resultant topoisomerase II-DNA complexes and DSBs account for the cytotoxic properties of the drugs. Topoisomerase poisons such as etoposide, epirubicin, mitoxantrone and mAMSA are of great clinical importance and are widely used in cancer therapy. Stabilised covalent topoisomerase II-DNA complexes can be repaired by the cell in a process that usually ends in the non homologous end joining (NHEJ) DNA double-strand break pathway. We are investigating the steps required before NHEJ can reseal the topoisomerase-induced break, using amongst others the TARDIS assay to quantify topoisomerase adducts in genomic DNA (14)
Changes in chromatin and nuclear organization associated with DNA damage and genome stability. The histone variant H2A.X is
rapidly
phosphorylated after cellular exposure to DNA-damaging agents such as
ionizing radiation or the topoisomerase poison etoposide, resulting in
chromatin domains enriched for
phosphorylated H2A.X (gammaH2AX) around sites of double-strand DNA
breaks. I previously showed that heterochromatin is a barrier to H2AX
phosphorylation (10) and work subsequently
published in Molecular Cell and Nature has confirmed this, showing that
removal of the heterochromatin protein HP1 is a necessary part of the
DNA damage response. Histone deacetylase inhibitors (HDACIs) cause
histone hyperacetylation and alter the properties of heterochromatin
relieving the block to H2AX phosphorylation. DNA topoisomerase II
poisons such as etoposide are mainstream cytotoxic anticancer drugs,
and recently it has been shown that their cytotoxic effects can be
potentiated by HDACIs. In Professor Austin's laboratory we are
interested in the interactions between histone deacetylase inhibitors,
DNA topoisomerase II poisons and heterochromatin with the aim of better
understanding the action of this class of drugs.We have recently shown that HDACIs cause a redistribution of topoisomerase II beta from heterochromatin to euchromatin in mouse cells and a change in the genomic distribution topoisomerase-mediated DNA damage (13). Targeting DNA-repair proteins as a means to improve the efficacy of anti-cancer treatments. I contributed to work validating the PIKK kinases
DNA-PKcs and ATM as drug targets for anti-cancer therapies (7,8 & 12) and with Drs Durkacz and Wilmore in the NICR
at Newcastle on an LLR-funded project to determine the role of DNA
damage-inducible kinases in the cellular responses to nucleoside
analogues used in leukaemia therapy (12). Histone lysine methylation and the function of chromodomain proteins. Work I started at
Newcastle and then continued at the Babraham and the Roslin Institutes
focused on histone modifications and the function of chromodomain
proteins (2-4). Chromodomain proteins,
particularly the heterochromatin protein HP1 bind to, and form the
“readout” for modifications including trimethylation of
histone H3 at lysine 9. I was amongst the first to report the
evolutionarily widespread occurrence of chromatin domains enriched for
histone H3 trimethylated at lysine 9, and the association of this
epigenetic modification with heterochromatin and a transcriptionally
silent state (5). Current work includes studying
the relationship between the epigenetic status of genomic regions and
their response to DNA damage induced by agents such as topoisomerase II
poisons or ionizing radiation (13) (see above).
|