5 EMIM: Input files
NOTE: EMIM is designed for the analysis of diallelic autosomal loci (i.e. SNPs on chromosomes 1-22) only. Please make sure to remove any non-autosomal or multi-allelic loci from your input files before running PREMIM and EMIM.
EMIM requires two compulsory input files, emimmarkers.dat and emimparams.dat, together with at least one out of three optional input data files (caseparenttrios.dat, casemotherduos.dat, casefatherduos.dat) and any number of additional optional input data files (caseparents.dat, casemothers.dat, casefathers.dat, cases.dat, conparents.dat, conmotherduos.dat, confatherduos.dat, cons.dat).
In previous versions of EMIM, these optional input data files had be created manually by the user, but now you can use PREMIM to generate these files automatically from standard PLINK format files. However, you may prefer to consider creating the file emimmarkers.dat yourself using the correct allele frequencies for your population (as PREMIM will use the observed allele frequencies in your sample, which may not be truly representative of population control frequencies). Alternatively, you could run PREMIM on some population control data (e.g. HapMap or WTCCC) to generate emimmarkers.dat, before running PREMIM on your own data to create the other files.
For most EMIM analyses the marker allele frequencies are used only as starting values for the numerical maximization procedure, and so it is not important that they are too accurate (as they will be re-estimated as required by EMIM). If your data consists of anything other than complete case/parent trios, then these estimated allele frequencies and/or mating type frequencies will influence the final results, in particular rendering the results potentially sensitive to population stratification. (See Ainsworth et al. (2011) for more details). For this reason, we recommend that you aim to ensure that your data set consists of individuals who are well matched for ancestry and come from a single homogeneous population.
If you prefer to create the EMIM input files manually yourself, you will have to decide on an allele coding scheme (alleles=1 and 2) and count up the number of individuals/trios/pairs with the various genotype combinations (genotypes 22, 12, 11) as required in the various EMIM input files described below.
WARNING!!!! A common problem sometimes encountered with the input files is when the final line of the file does not have a newline character. If you get an error message such as “fortran runtime error: End of file” please check that all your input files have a newline character at the end of the final line.
emimmarkers.dat: This file lists on each line (in order) a numeric SNP ID (which could correspond to SNP number or base pair position, for example) for each SNP to be analysed, together with the allele frequency for the allele denoted “2” (usually the minor allele) at each SNP.
For example, if there are 8 different SNPs to be analysed, with SNPs 1 to 8 having minor allele frequencies 0.419927, 0.28163, 0.01, 0.152068, 0.265815, 0.0991484, 0.129562 and 0.379584 respetively, emimmarkers.dat might look like:
1 0.419927
2 0.28163
3 0.01
4 0.152068
5 0.265815
6 0.0991484
7 0.129562
8 0.379584
The allele frequencies are generally only used as starting values (unless you choose the option in EMIM to fix the allele frequency at its starting value - which is NOT recommended). Therefore the allele frequencies do not have to be too accurate. An estimate from HapMap, or from previous genetic studies in your population, or even estimated from your own data set (using PREMIM) should be sufficient.
emimparams.dat: This is a file that tells EMIM what input files to read in and sets up the various parameter restrictions for the analyses to be performed. An example of this file is shown on the following page. Here we describe the lines of this file in detail:
Lines 1, 14 and 34 are not used by the program but are simply separators to make the 3 sections of this file easier to read. The text after  on each other line of the file is not read by the program, but is designed to describe what the number (1 or 0) at the beginning of the line means. You are strongly recommended to keep these text comments in order to avoid mistakes. The order of the lines must be EXACTLY as shown in this example.
on each other line of the file is not read by the program, but is designed to describe what the number (1 or 0) at the beginning of the line means. You are strongly recommended to keep these text comments in order to avoid mistakes. The order of the lines must be EXACTLY as shown in this example.
Lines 2-12 tell EMIM what input files to expect (note that the names given to these input files are not optional). A “1” indicates that this input file exists and is to be read in, while a “0” indicates that this input file is not to be read in. At least one of the files caseparenttrios.dat, casemotherduos.dat, casefatherduos.dat (lines 2, 4 and 5) must be read in; in the example above all three of these files are read in, as well as a conmotherduos.dat file.
Line 13 tells EMIM how many SNPs to be analysed. Normally this number will match the number of SNPs in the file emimmarkers.dat. If this number n is less than the number of SNPs in the file emimmarkers.dat, then only the first n SNPs in the file emimmarkers.dat will be analysed. If this number n is greater than the number of SNPs in the file emimmarkers.dat, then EMIM will simply stop after it has analysed all the SNPs listed in emimmarkers.dat
The second and third sections of the file have a number of lines telling what parameters to estimate and what parameter restrictions to use. A “1” indicates that this parameter is to be estimated or this restriction is to be used. A “0” indicates that this parameter is not to be estimated or this restriction is not to be used.
-----------INPUT DATAFILES------------------------------------------------
1 << caseparenttrios.dat file (0=no, 1=yes)
1 << caseparents.dat file (0=no, 1=yes)
1 << casemotherduos.dat file (0=no, 1=yes)
0 << casefatherduos.dat file (0=no, 1=yes)
1 << casemothers.dat file (0=no, 1=yes)
0 << casefathers.dat file (0=no, 1=yes)
1 << cases.dat file (0=no, 1=yes)
1 << conparents.dat file (0=no, 1=yes)
0 << conmotherduos.dat file (0=no, 1=yes)
0 << confatherduos.dat file (0=no, 1=yes)
1 << cons.dat file (0=no, 1=yes)
634 << no of SNPs in each file
------------------PARAMETER RESTRICTIONS----------------------------------
0 << fix allele freq A (0=no, 1=yes)
1 << assume HWE and random mating (0=no=estimate 6 mu parameters, 1=yes)
0 << assume parental allelic exchangeability (0=no, 1=yes)
0 << use CPG likelihood (estimate 9 mu parameters)
1 << estimate R1 (0=no, 1=yes)
1 << estimate R2 (0=no, 1=yes)
0 << R2=R1 (0=no, 1=yes)
0 << R2=R1squared (0=no, 1=yes)
0 << estimate S1 (0=no, 1=yes)
0 << estimate S2 (0=no, 1=yes)
0 << S2=S1 (0=no, 1=yes)
0 << S2=S1squared (0=no, 1=yes)
1 << estimate Im (0=no, 1=yes)
0 << estimate Ip (0=no, 1=yes)
0 << estimate gamma11 (0=no, 1=yes)
0 << estimate gamma12 (0=no, 1=yes)
0 << estimate gamma21 (0=no, 1=yes)
0 << estimate gamma22 (0=no, 1=yes)
0 << gamma22=gamma12= gamma21=gamma11 (0=no, 1=yes)
---------------OTHER PARAMETERIZATIONS------------------------------------
0 << estimate Weinberg (1999b) Im (0=no, 1=yes)
0 << estimate Weinberg (1999b) Ip (=Li 2009 Jm) (0=no, 1=yes)
0 << estimate Sinsheimer (2003) gamma01 (0=no, 1=yes)
0 << estimate Sinsheimer (2003) gamma21 (0=no, 1=yes)
0 << estimate Palmer (2006) match parameter (0=no, 1=yes)
0 << estimate Li (2009) conflict parameter Jc (0=no, 1=yes)
Line 15 << fix allele freq A (0=no, 1=yes) indicates that the allele frequencies are to be fixed at their given starting values (NOT RECOMMENDED). A “1” in this line will supercede any instructions given in the next two lines (lines 16 and 17).
Line 16 << assume HWE and random mating (0=no=estimate 6 mu parameters, 1=yes) indicates that the analysis should be performed assuming Hardy Weinberg Equilibrium (HWE) and random mating. In that case, one allele frequency parameter 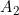 (the frequency of the 2 allele) will be estimated (or fixed) as opposed to estimating six mating-type stratification parameters
(the frequency of the 2 allele) will be estimated (or fixed) as opposed to estimating six mating-type stratification parameters 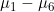 . A “1” in this line will supercede any instructions given in the next line (line 17).
. A “1” in this line will supercede any instructions given in the next line (line 17).
Line 17 << assume parental allelic exchangeability (0=no, 1=yes) indicates that parental allelic exchangeability should be assumed (i.e. 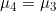 ) when estimating .
) when estimating .
Line 18 << use CPG likelihood (estimate 9 mu parameters) indicates that the Conditional on Parental Genotypes (CPG) rather than the Conditional on Exchangeable Parental Genotypes (CEPG) [Cordell (2004), Weinberg and Shi (2009)] likelihood should be used. This provides a more robust analysis that does not assume mating symmetry (exchangeability of parental mating types), at the expense of estimating a larger number (nine) of mating-type stratification parameters. This analysis is recommended if your derives from pedigrees that contain (or were ascertained on the basis of the presence of) multiple affected individuals [Cordell (2004)].
Line 19 << estimate R1 (0=no, 1=yes) indicates that the child genotype effect 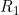 (the factor by which the disease risk is multiplied if the child has a single copy of allele 2) should be estimated.
(the factor by which the disease risk is multiplied if the child has a single copy of allele 2) should be estimated.
Line 20 << estimate R2 (0=no, 1=yes) indicates that the child genotype effect 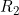 (the factor by which the disease risk is multiplied if the child has two copies of allele 2) should be estimated.
(the factor by which the disease risk is multiplied if the child has two copies of allele 2) should be estimated.
Line 21 << R2=R1 (0=no, 1=yes) indicates that a single child genotype effect 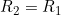 should be estimated. A “1” in this line will supercede any instructions given in the two previous lines. However, if line 21 is set equal to “1”, we recommend you set lines 19 and 20 to “0” in order to avoid problems when EMIM tries to determine whether the parameters you have selected are estimable, given the data.
should be estimated. A “1” in this line will supercede any instructions given in the two previous lines. However, if line 21 is set equal to “1”, we recommend you set lines 19 and 20 to “0” in order to avoid problems when EMIM tries to determine whether the parameters you have selected are estimable, given the data.
Line 22 << R2=R1squared (0=no, 1=yes) indicates that a single child genotype effect 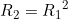 should be estimated. This is a multiplicative allelic model for the child genotype effects. A “1” in this line will supercede any instructions given in the three previous lines. However, if line 22 is set equal to “1”, we recommend you set lines 19, 20 and 21 to “0” in order to avoid problems when EMIM tries to determine whether the parameters you have selected are estimable, given the data.
should be estimated. This is a multiplicative allelic model for the child genotype effects. A “1” in this line will supercede any instructions given in the three previous lines. However, if line 22 is set equal to “1”, we recommend you set lines 19, 20 and 21 to “0” in order to avoid problems when EMIM tries to determine whether the parameters you have selected are estimable, given the data.
Line 23 << estimate S1 (0=no, 1=yes) indicates that the maternal genotype effect 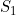 (the factor by which the disease risk is multiplied if the mother has a single copy of allele 2) should be estimated.
(the factor by which the disease risk is multiplied if the mother has a single copy of allele 2) should be estimated.
Line 24 << estimate S2 (0=no, 1=yes) indicates that the maternal genotype effect 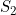 (the factor by which the disease risk is multiplied if the mother has two copies of allele 2) should be estimated.
(the factor by which the disease risk is multiplied if the mother has two copies of allele 2) should be estimated.
Line 25 << S2=S1 (0=no, 1=yes) indicates that a single maternal genotype effect 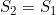 should be estimated. A “1” in this line will supercede any instructions given in the two previous lines. However, if line 25 is set equal to “1”, we recommend you set lines 23 and 24 to “0” in order to avoid problems when EMIM tries to determine whether the parameters you have selected are estimable, given the data.
should be estimated. A “1” in this line will supercede any instructions given in the two previous lines. However, if line 25 is set equal to “1”, we recommend you set lines 23 and 24 to “0” in order to avoid problems when EMIM tries to determine whether the parameters you have selected are estimable, given the data.
Line 26 << S2=S1squared (0=no, 1=yes) indicates that a single maternal genotype effect 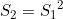 should be estimated. This is a multiplicative allelic model for the maternal genotype effects. A “1” in this line will supercede any instructions given in the three previous lines. However, if line 26 is set equal to “1”, we recommend you set lines 23, 24 and 25 to “0” in order to avoid problems when EMIM tries to determine whether the parameters you have selected are estimable, given the data.
should be estimated. This is a multiplicative allelic model for the maternal genotype effects. A “1” in this line will supercede any instructions given in the three previous lines. However, if line 26 is set equal to “1”, we recommend you set lines 23, 24 and 25 to “0” in order to avoid problems when EMIM tries to determine whether the parameters you have selected are estimable, given the data.
Line 27 << estimate Im (0=no, 1=yes) indicates that a maternal imprinting effect 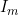 (a multiplicative factor by which the probability of disease is multiplied if the child receives a (maternal) copy of the 2 allele from their mother) should be estimated. A “1” in this line will supercede any instructions given in the next line (line 28) i.e. only one of and
(a multiplicative factor by which the probability of disease is multiplied if the child receives a (maternal) copy of the 2 allele from their mother) should be estimated. A “1” in this line will supercede any instructions given in the next line (line 28) i.e. only one of and 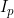 can be estimated. The exception to this is if no child genotype (R) or interaction (gamma) parameters are estimated, in which case it is possible to estimate both and .
can be estimated. The exception to this is if no child genotype (R) or interaction (gamma) parameters are estimated, in which case it is possible to estimate both and .
Line 28 << estimate Ip (0=no, 1=yes) indicates that a paternal imprinting effect (a multiplicative factor by which the probability of disease is multiplied if the child receives a (paternal) copy of the 2 allele from their father) should be estimated.
Line 29 << estimate gamma11 (0=no, 1=yes) indicates that the mother/child genotype interaction parameter 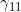 should be estimated.
should be estimated.
Line 30 << estimate gamma12 (0=no, 1=yes) indicates that the mother/child genotype interaction parameter  should be estimated.
should be estimated.
Line 31 << estimate gamma21 (0=no, 1=yes) indicates that the mother/child genotype interaction parameter 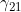 should be estimated.
should be estimated.
Line 32 << estimate gamma22 (0=no, 1=yes) indicates that the mother/child genotype interaction parameter 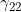 should be estimated.
should be estimated.
Line 33 << gamma22=gamma12=gamma21=gamma11 (0=no, 1=yes) indicates that a single mother/child genotype interaction parameter 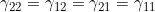 should be estimated. A “1” in this line will supercede any instructions given in the four previous lines. However, if line 33 is set equal to “1”, we recommend you set lines 29, 30, 31 and 32 to “0” in order to avoid problems when EMIM tries to determine whether the parameters you have selected are estimable, given the data.
should be estimated. A “1” in this line will supercede any instructions given in the four previous lines. However, if line 33 is set equal to “1”, we recommend you set lines 29, 30, 31 and 32 to “0” in order to avoid problems when EMIM tries to determine whether the parameters you have selected are estimable, given the data.
Depending on what optional input data files are available, estimation of certain parameter combinations may be limited. (This is particularly true if you only read in a single file, casemotherduos.dat or casefatherduos.dat ). EMIM will attempt to adjust the number of parameters to estimate in some “sensible” way if it detects you are trying to estimate too many parameters with not enough restrictions. However, it may be better to make this adjustment yourself (e.g. by making assumptions of HWE and/or estimating only a smaller number of parameters). You can generally tell if EMIM has been successful at its choice of parameters by looking at the output confidence intervals: if these do not look sensible (e.g. if the upper and lower confidence limits for a parameter are equal) then there is a good chance that the choice of parameters has not been made appropriately.
Lines 35 and 36 << estimate Weinberg (1999b) Im (0=no, 1=yes) << estimate Weinberg (1999b) Ip (=Li 2009 Jm) (0=no, 1=yes)
Parameterization of interactions and imprinting effects is quite complex (see Ainsworth et al. (2011)) and several different parameterizations have been proposed in the literature. Our parameterization for the parent-of-origin effects and corresponds to the original parameterization used by Weinberg et al. (1998) rather than to a later alternative parameterization used by Weinberg (1999), Parimi et al. (2008), and Li et al. (2009). If preferred, the user can choose to use the later parameterization by setting the values in lines 27 and 28 to 0 and the values in line 35 or 36 to 1. In this case, if interactions are also required, we recommend using either the Sinsheimer et al. (2003) or Palmer et al. (2006) parameterization (see below), as our interaction parameterization does not allow estimation of the later Weinberg (1999) imprinting parameters.
Lines 37 and 38 << estimate Sinsheimer (2003) gamma01 (0=no, 1=yes) << estimate Sinsheimer (2003) gamma21 (0=no, 1=yes) Sinsheimer et al. (2003) proposed an alternative parameterization for interactions in terms of maternal-fetal genotype incompatibility (MFG) parameters. Sinsheimer et al. (2003) denoted these parameters as 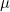 (or
(or 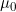 ) and
) and 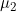 . We denote these MFG interactions as
. We denote these MFG interactions as 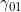 and , since they correspond to effects that operate (in addition to maternal and child genotype effects) when the child has one copy, and the mother either zero or two copies, of a particular allele of interest. To include one or both MFG interactions, you should set the values in lines 29-33 to 0 and the value(s) in line 37 and/or 38 to 1.
and , since they correspond to effects that operate (in addition to maternal and child genotype effects) when the child has one copy, and the mother either zero or two copies, of a particular allele of interest. To include one or both MFG interactions, you should set the values in lines 29-33 to 0 and the value(s) in line 37 and/or 38 to 1.
Line 39 << estimate Palmer (2006) match parameter (0=no, 1=yes) Sinsheimer et al. (2003) and Palmer et al. (2006) considered an alternative interaction parameterization in which “matching” rather “mismatching” between maternal and fetal genotypes increases disease risk in the offspring. To model interaction via the single Palmer et al. (2006) match parameter , you should set the values in lines 29-33 to 0 and the value in line 39 to 1.
Line 40 << estimate Li (2009) conflict parameter Jc (0=no, 1=yes) Li et al. (2009) (based on work by Parimi et al. (2008)) considered an alternative interaction parameterization that modelled “conflict” between the mothers and childs genotypes. To model interaction via the single Li et al. (2009) conflict parameter (which we denote 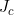 , corresponding to exp(
, corresponding to exp(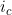 ) in the notation of Li et al. (2009)), you should set the values in lines 29-33 to 0 and the value in line 40 to 1. Note that the recommended model of Li et al. (2009) and Parimi et al. (2008)) is to include both and
) in the notation of Li et al. (2009)), you should set the values in lines 29-33 to 0 and the value in line 40 to 1. Note that the recommended model of Li et al. (2009) and Parimi et al. (2008)) is to include both and 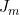 (=exp(
(=exp(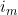 ) in the notation of Li et al. (2009)) where is the imprinting parameter selectable on line 36. So to fit the full Li et al. (2009) and Parimi et al. (2008) model you should set the values in lines 36 and 40 to 1.
) in the notation of Li et al. (2009)) where is the imprinting parameter selectable on line 36. So to fit the full Li et al. (2009) and Parimi et al. (2008) model you should set the values in lines 36 and 40 to 1.
caseparenttrios.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the 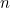 SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 15 cell counts corresponding to the number of fully genotyped case/parent trios whose genotype combinations fall into the appropriate genotype categories as given in Ainsworth et al. (2011) Table 1. (Zero counts are allowed, although may make it more difficult to estimate certain parameter combinations).
For example, suppose that at the first SNP the genotype combinations of mother, father and child as given in Ainsworth et al. (2011) Table 1 are:
| group | mother | father | child | count |
|---|
| 1 | 22 | 22 | 22 | 4 |
| 2 | 22 | 12 | 22 | 10 |
| 3 | 22 | 12 | 12 | 17 |
| 4 | 12 | 22 | 22 | 9 |
| 5 | 12 | 22 | 12 | 13 |
| 6 | 22 | 11 | 12 | 6 |
| 7 | 11 | 22 | 12 | 4 |
| 8 | 12 | 12 | 22 | 14 |
| 9 | 12 | 12 | 12 | 44 |
| 10 | 12 | 12 | 11 | 26 |
| 11 | 12 | 11 | 12 | 25 |
| 12 | 12 | 11 | 11 | 17 |
| 13 | 11 | 12 | 12 | 24 |
| 14 | 11 | 12 | 11 | 12 |
| 15 | 11 | 11 | 11 | 19 |
Then the line in caseparenttrios.dat corresponding to this SNP would look like:
1 4 10 17 9 13 6 4 14 44 26 25 17 24 12 19
An example of caseparenttrios.dat for 8 SNPs, of which the first has counts as given above, is shown below:
snp cellcount 1-15
1 4 10 17 9 13 6 4 14 44 26 25 17 24 12 19
2 1 0 8 0 5 2 3 2 23 21 29 38 26 43 52
3 0 0 0 0 0 0 0 0 0 1 1 0 1 0 319
4 0 0 3 0 0 2 4 1 13 24 22 40 8 25 131
5 0 0 5 3 6 4 7 2 23 23 29 44 19 46 59
6 0 0 1 2 0 0 2 2 4 8 14 18 21 26 197
7 0 0 3 0 0 1 0 1 11 18 14 37 12 29 160
8 1 3 9 5 12 9 8 12 31 30 28 26 31 20 20
caseparents.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 9 cell counts corresponding to the number of fully genotyped parents of cases whose genotype combinations fall into the appropriate genotype categories. Note that these parents of cases must not include parents of cases who have already appeared as case/parent trios in the file caseparenttrios.dat (i.e. all input data files must be independent).
For example, suppose that at the first SNP the genotype combinations of the mother and father are
| group | mother | father | count |
|---|
| 1 | 22 | 22 | 0 |
| 2 | 22 | 12 | 0 |
| 3 | 22 | 11 | 1 |
| 4 | 12 | 22 | 0 |
| 5 | 12 | 12 | 0 |
| 6 | 12 | 11 | 0 |
| 7 | 11 | 22 | 0 |
| 8 | 11 | 12 | 2 |
| 9 | 11 | 11 | 16 |
Then the line in caseparents.dat corresponding to this SNP would look like:
1 0 0 1 0 0 0 0 2 16
An example of caseparents.dat for 8 SNPs, of which the first has counts as given above, is shown below:
snp cellcount 1-9
1 0 0 1 0 0 0 0 2 16
2 0 0 0 0 1 3 0 1 9
3 0 0 0 0 0 0 0 0 13
4 0 0 1 0 0 0 0 0 1
5 0 0 0 0 1 0 0 1 0
6 0 0 0 0 0 0 0 0 0
7 0 0 0 0 0 0 1 0 0
8 0 0 1 0 0 1 0 3 0
casemotherduos.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 7 cell counts corresponding to the number of fully genotyped case/mother duos whose genotype combinations fall into the appropriate genotype categories. Note that these must not include cases and mothers who have already appeared as case/parent trios in the file caseparenttrios.dat, or mothers who have already appeared in the file caseparents.dat (i.e. all input data files must be independent). Zero counts are allowed, although may make it more difficult to estimate certain parameter combinations.
For example, suppose that at the first SNP the genotype combinations of the mother and child are
| group | mother | child | count |
|---|
| 1 | 22 | 22 | 4 |
| 2 | 22 | 12 | 3 |
| 3 | 12 | 22 | 1 |
| 4 | 12 | 12 | 7 |
| 5 | 12 | 11 | 4 |
| 6 | 11 | 12 | 3 |
| 7 | 11 | 11 | 4 |
Then the line in casemotherduos.dat corresponding to this SNP would look like:
1 4 3 1 7 4 3 4
An example of casemotherduos.dat for 8 SNPs, of which the first has counts as given above, is shown below:
snp cellcount 1-7
1 4 3 1 7 4 3 4
2 0 1 3 3 6 5 7
3 0 1 0 0 0 0 25
4 0 0 0 2 3 5 15
5 3 1 1 8 2 5 4
6 0 0 1 2 4 2 17
7 0 0 0 2 2 5 17
8 1 3 3 6 4 0 7
casefatherduos.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 7 cell counts corresponding to the number of fully genotyped case/father duos whose genotype combinations fall into the appropriate genotype categories. Note that these must not include cases and fathers who have already appeared as case/parent trios in the file caseparenttrios.dat, fathers who have already appeared in the file caseparents.dat, or cases who have already appeared in the file casemotherduos.dat (i.e. all input data files must be independent). Zero counts are allowed, although may make it more difficult to estimate certain parameter combinations.
For example, suppose that at the first SNP the genotype combinations of the father and child are
| group | father | child | count |
|---|
| 1 | 22 | 22 | 1 |
| 2 | 22 | 12 | 0 |
| 3 | 12 | 22 | 2 |
| 4 | 12 | 12 | 1 |
| 5 | 12 | 11 | 2 |
| 6 | 11 | 12 | 3 |
| 7 | 11 | 11 | 3 |
Then the line in casefatherduos.dat corresponding to this SNP would look like:
1 1 0 2 1 2 3 3
An example of casefatherduos.dat for 8 SNPs, of which the first has counts as given above, is shown below:
snp cellcount 1-7
1 1 0 2 1 2 3 3
2 0 0 1 0 5 4 1
3 0 0 0 0 0 0 11
4 0 1 0 3 0 0 7
5 0 2 1 3 1 2 2
6 0 0 0 0 2 3 6
7 0 0 0 0 0 3 8
8 1 0 1 2 2 2 5
casemothers.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 3 cell counts corresponding to the number of fully genotyped mothers of cases whose genotypes fall into the appropriate genotype categories. Note that these must not include mothers of cases already in the files caseparenttrios.dat, caseparents.dat, or casemotherduos.dat (i.e. all input data files must be independent).
For example, suppose that at the first SNP the genotypes of the mothers are
| group | mother | count |
|---|
| 1 | 22 | 18 |
| 2 | 12 | 160 |
| 3 | 11 | 358 |
Then the line in casemothers.dat corresponding to this SNP would look like:
1 18 160 358
An example of casemothers.dat for 8 SNPs, of which the first has counts as given above, is shown below:
snp cellcount 1-3
1 18 160 358
2 8 90 438
3 5 85 445
4 8 147 375
5 4 97 436
6 1 32 503
7 30 157 349
8 5 64 467
casefathers.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 3 cell counts corresponding to the number of fully genotyped fathers of cases whose genotypes fall into the appropriate genotype categories. Note that these must not include fathers of cases already in the files caseparenttrios.dat, caseparents.dat, or casefatherduos.dat (i.e. all input data files must be independent).
For example, suppose that at the first SNP the genotypes of the fathers are
| group | father | count |
|---|
| 1 | 22 | 4 |
| 2 | 12 | 71 |
| 3 | 11 | 461 |
Then the line in casefathers.dat corresponding to this SNP would look like:
1 4 71 461
An example of casefathers.dat for 8 SNPs, of which the first has counts as given above, is shown below:
snp cellcount 1-3
1 4 71 461
2 5 64 467
3 2 27 506
4 82 239 214
5 4 55 477
6 21 162 353
7 4 62 470
8 11 110 415
cases.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 3 cell counts corresponding to the number of fully genotyped cases whose genotypes fall into the appropriate genotype categories. Note that these must not include cases already in the files caseparenttrios.dat, casemotherduos.dat, or casefatherduos.dat (i.e. all input data files must be independent).
For example, suppose that at the first SNP the genotypes of the cases are
| group | case | count |
|---|
| 1 | 22 | 93 |
| 2 | 12 | 268 |
| 3 | 11 | 174 |
Then the line in cases.dat corresponding to this SNP would look like:
1 93 268 174
An example of cases.dat for 8 SNPs, of which the first has counts as given above, is shown below:
snp cellcount 1-3
1 93 268 174
2 47 223 265
3 0 0 536
4 11 150 375
5 51 197 288
6 7 93 436
7 9 128 399
8 99 247 189
conparents.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 9 cell counts corresponding to the number of fully genotyped parents of controls whose genotype combinations fall into the appropriate genotype categories.
Note that by controls we mean individuals of unknown disease status, or (provided the disease is rare) individuals who are known to be disease-free. Note that these parents of controls must not include parents who have already appeared in any other input files (i.e. all input data files must be independent).
For example, suppose that at the first SNP the genotype combinations of the mother and father are
| group | mother | father | count |
|---|
| 1 | 22 | 22 | 0 |
| 2 | 22 | 12 | 0 |
| 3 | 22 | 11 | 0 |
| 4 | 12 | 22 | 0 |
| 5 | 12 | 12 | 0 |
| 6 | 12 | 11 | 0 |
| 7 | 11 | 22 | 0 |
| 8 | 11 | 12 | 1 |
| 9 | 11 | 11 | 0 |
Then the line in conparents.dat corresponding to this SNP would look like:
1 0 0 0 0 0 0 0 1 0
An example of conparents.dat for 8 SNPs, of which the first has counts as given above, is shown below:
snp cellcount 1-9
1 0 0 0 0 0 0 0 1 0
2 0 0 0 0 0 0 0 0 1
3 0 0 0 0 0 0 0 0 1
4 0 0 0 0 1 0 0 0 0
5 0 0 0 0 0 0 0 1 0
6 0 0 0 0 1 0 0 0 0
7 0 0 0 0 0 0 0 1 0
8 0 0 0 0 0 1 0 0 0
conmotherduos.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 7 cell counts corresponding to the number of fully genotyped control/mother duos whose genotype combinations fall into the appropriate genotype categories. Note that by controls we mean individuals of unknown disease status, or (provided the disease is rare) individuals who are known to be disease-free. This must not include individuals who have already appeared in any other input files (i.e. all input data files must be independent).
For example, suppose that at the first SNP the genotype combinations of the mother and child are
| group | mother | child | count |
|---|
| 1 | 22 | 22 | 0 |
| 2 | 22 | 12 | 0 |
| 3 | 12 | 22 | 1 |
| 4 | 12 | 12 | 1 |
| 5 | 12 | 11 | 2 |
| 6 | 11 | 12 | 2 |
| 7 | 11 | 11 | 21 |
Then the line in conmotherduos.dat corresponding to this SNP would look like:
1 0 0 1 1 2 2 21
An example of conmotherduos.dat for 8 SNPs, of which the first has counts as given above, is shown below:
snp cellcount 1-7
1 0 0 1 1 2 2 21
2 0 0 0 0 3 7 16
3 0 0 0 1 2 2 20
4 0 0 0 2 4 2 16
5 0 0 2 1 2 1 19
6 0 0 0 1 2 0 23
7 0 0 0 3 5 5 13
8 0 0 0 1 1 3 21
confatherduos.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 7 cell counts corresponding to the number of fully genotyped control/father duos whose genotype combinations fall into the appropriate genotype categories. Note that by controls we mean individuals of unknown disease status, or (provided the disease is rare) individuals who are known to be disease-free. This must not include individuals who have already appeared in any other input files (i.e. all input data files must be independent).
For example, suppose that at the first SNP the genotype combinations of the father and child are
| group | father | child | count |
|---|
| 1 | 22 | 22 | 0 |
| 2 | 22 | 12 | 1 |
| 3 | 12 | 22 | 0 |
| 4 | 12 | 12 | 1 |
| 5 | 12 | 11 | 1 |
| 6 | 11 | 12 | 2 |
| 7 | 11 | 11 | 6 |
Then the line in confatherduos.dat corresponding to this SNP would look like:
1 0 1 0 1 1 2 6
An example of confatherduos.dat for 8 SNPs, of which the first has counts as given above, and the other seven just happen to all have exactly the same (different set of) genotype counts (admittedly a contrived example is shown below:
snp cellcount 1-7
1 0 1 0 1 1 2 6
2 0 0 0 0 1 2 8
3 0 0 0 0 3 1 7
4 0 0 0 0 5 3 3
5 0 0 0 3 1 0 7
6 0 0 0 0 1 0 10
7 0 0 0 0 2 4 5
8 0 1 0 0 0 0 10
cons.dat: This file contains a header line (which is not used by the program but is useful for reminding yourself of the column order), followed by a line of data for each of the SNPs to be analysed (IN EXACTLY THE SAME ORDER as given in emimmarkers.dat).
The first number on each line is the numeric SNP ID (as given in emimmarkers.dat). This is followed by 3 cell counts corresponding to the number of fully genotyped controls whose genotypes fall into the appropriate genotype categories. Note that by controls we mean individuals of unknown disease status, or (provided the disease is rare) individuals who are known to be disease-free. This must not include individuals who have already appeared in any other input files (i.e. all input data files must be independent).
For example, suppose that at the first SNP the genotypes of the controls are
| group | control | count |
|---|
| 1 | 22 | 1 |
| 2 | 12 | 7 |
| 3 | 11 | 6 |
Then the line in cons.dat corresponding to this SNP would look like:
1 1 7 6
An example of cons.dat for 8 SNPs, of which the first has counts as given above, is shown below:
snp cellcount 1-3
1 1 7 6
2 1 4 9
3 1 1 12
4 0 1 13
5 1 5 8
6 0 7 7
7 0 2 12
8 0 5 9
