6 EMIM: Usage and output files
The program is run by typing “./emim”. This should produce 2 primary output files: emimresults.out and emimsummary.out.
In previous versions of EMIM, two additional files (fort.11 and fort.12) with details of the maximisation procedure were also produced by the MAXFUN subroutine. These files can be quite large and may subsequently be deleted if detailed output is not required. (However they can be quite useful for troubleshooting when the maximisation procedure does not appear to have worked correctly). In current versions of EMIM we do not output these files by default. Please contact us if you need a version of EMIM that outputs these files.
The main results are given in the file emimresults.out. A fairly large number of lines of results are output for each SNP in turn, therefore this file can become quite large. If you particularly want to see the detailed results for some specific SNPs, we recommend you use PLINK to create a smaller subset containing just the SNPs of interest, and then re-run PREMIM and EMIM on this subset.
The file emimresults.out first shows the parameter estimates from a global null model (in which the parameters 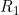 ,
, 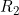 ,
, 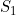 ,
, 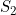 ,
, 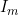 ,
, 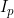 ,
, 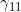 ,
,  ,
, 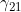 ,
, 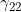 are all set to equal 1.0). Next the parameter estimates from the specified alternative model (as specified in the file emimparams.dat) are given, both on their original and log scales, followed by 95% confidence intervals for estimated parameters of interest. Finally the maximized ln likelihoods for the alternative and null models are given, together with twice the difference between these (which can be compared to a chi-squared on the appropriate df (=the number of estimated parameters of interest) to calculate a p-value, if required). Note that if nested alternative (non-null) models are to be compared, you will have to run EMIM twice and compare twice the difference between the maximized ln likelihoods for each of the alternative models to a chi-squared on the appropriate df.
are all set to equal 1.0). Next the parameter estimates from the specified alternative model (as specified in the file emimparams.dat) are given, both on their original and log scales, followed by 95% confidence intervals for estimated parameters of interest. Finally the maximized ln likelihoods for the alternative and null models are given, together with twice the difference between these (which can be compared to a chi-squared on the appropriate df (=the number of estimated parameters of interest) to calculate a p-value, if required). Note that if nested alternative (non-null) models are to be compared, you will have to run EMIM twice and compare twice the difference between the maximized ln likelihoods for each of the alternative models to a chi-squared on the appropriate df.
A summary of the results from emimresults.out is given in the file emimsummary.out. This file can be more convenient to deal with (e.g. by reading in and manipulating in R) if analysis is being performed on a large number of SNPs. First comes a header line describing the different columns. Then comes a single line of results for each SNP analysed. The entries in each line correspond to the SNP number, the SNP ID, the estimated allele frequency (or input allele frequency if allele frequency is not estimated), then for each parameter (, , , , , , , , , ) in turn, we have the estimate of the logarithm of the relevant parameter, its estimated standard error, and the estimated lower and upper 95% confidence limit for the logarithm of the relevant parameter. Finally we have the maximized log likelihood under the null, the maximized log likelihood under the alternative, and twice the difference between these, followed by a column indicating whether there were any warning messages for this SNP output to the file emimresults.out (in which case we recommend you take a look at the file emimresults.out to see what the warning messages say).
PLEASE NOTE THAT EMIM DOES NOT ITSELF GENERATE ANY P-VALUES. Instead, EMIM produces chi-squared statistics and maximized log likelihoods (that can be compared for nested models and used to create p-values, see Example section below). In order to calculate the p-value for any test, you will therefore need to compare the chi-squared statistic or twice the difference in maximized log likelihoods to a chi-squared on the appropriate degrees of freedom, using an external program such as Excel or R.
If one of the parameterizations from the third section of emimparams.dat has been selected, the output parameters in emimsummary.out are slightly different. If the Sinsheimer et al. (2003) MFG interaction parameters have been chosen, then instead of and/or , you should find that MFG parameters 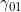 and/or (denoted MFG01 and MFG21) are output. If the Palmer et al. (2006) interaction parameter has been chosen, then instead of you should find the Palmer match parameter (denoted MFGmu) is output. If the Li et al. (2009) and Parimi et al. (2008) interaction parameter has been chosen, then instead of , you should find that the Li et al. (2009) conflict parameter (denoted Jc) is output. Hopefully the header line in emimsummary.out should make it clear which parameters have been output.
and/or (denoted MFG01 and MFG21) are output. If the Palmer et al. (2006) interaction parameter has been chosen, then instead of you should find the Palmer match parameter (denoted MFGmu) is output. If the Li et al. (2009) and Parimi et al. (2008) interaction parameter has been chosen, then instead of , you should find that the Li et al. (2009) conflict parameter (denoted Jc) is output. Hopefully the header line in emimsummary.out should make it clear which parameters have been output.
Moreover, if one of the parameterizations from the third section of emimparams.dat has been selected, the value of the other estimated parameters may be different. For example, if the Sinsheimer et al. (2003) MFG interaction parameterisation is chosen, the R1 and R2 parameters as output by EMIM will actually correspond to what were called 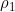 and
and 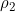 in Table I (Example 9) of Ainsworth et al. (2011), and and will actually correspond to what were called
in Table I (Example 9) of Ainsworth et al. (2011), and and will actually correspond to what were called  and
and 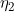 in Table I (Example 9) of Ainsworth et al. (2011). Perusal of Table I (Example 9) of Ainsworth et al. (2011) shows that () is the factor by which an individual's disease risk is multiplied if they possess one (two) risk alleles at a given locus, and () is the factor by which an individual's disease risk is multiplied if their mother possesses one risk allele at that locus, when using the Sinsheimer parameterisation. That is why these parameters are still denoted , , and in the EMIM output - because they have the same meaning that we are attributing to , , and when using the default interaction parameterisation (i.e. when interaction is modelled in terms of parameters and , instead of and ). However, the values of , , and will not necessarily be the same within these two paramaterisations. See Ainsworth et al. (2011) more discussion of this issue.
in Table I (Example 9) of Ainsworth et al. (2011). Perusal of Table I (Example 9) of Ainsworth et al. (2011) shows that () is the factor by which an individual's disease risk is multiplied if they possess one (two) risk alleles at a given locus, and () is the factor by which an individual's disease risk is multiplied if their mother possesses one risk allele at that locus, when using the Sinsheimer parameterisation. That is why these parameters are still denoted , , and in the EMIM output - because they have the same meaning that we are attributing to , , and when using the default interaction parameterisation (i.e. when interaction is modelled in terms of parameters and , instead of and ). However, the values of , , and will not necessarily be the same within these two paramaterisations. See Ainsworth et al. (2011) more discussion of this issue.
So, in summary, if using the Sinsheimer et al. (2003) MFG interaction parameterisation, the parameters / / / / / estimated by EMIM are actually equivalent to the parameters that were called / / / / 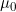 /
/ 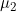 by Sinsheimer et al. (2003).
by Sinsheimer et al. (2003).
