10 Troubleshooting and results interpretation
PREMIM and EMIM are ideally designed for use by users who already have experience of analysing genetic data and some familiarity with using command-line programs such as PLINK, SNPTEST, MACH or IMPUTE. If you do not have any such experience, we recommend you attept to gain some familiarity with such programs before embarking on an analysis with PREMIM and EMIM.
As with any other statistical analysis method, the results from EMIM are only as reliable as the quality of the data going in to the analysis. If you obtain "strange" results or get a warning/error message, by far the most likely reason is that there is some "problem" with the input data. By "problem", this could mean an actual mistake in the input files (e.g. they are not formatted correctly), or it could simply mean that there is limited information provided by the input data, or that the data is too noisy, to produce reliable/interpretable results.
One common problem is that there is too little data to estimate the parameters requested. This could result from too many cells with zero observations in the input data files caseparenttrios.dat, casemotherduos.dat etc. We have tried to get EMIM to pick up these sorts of issues automatically and give a sensible warning message, but, given the complexities of the potential possible models, it does not always manage to achieve this! You may find that using a more restricted model (e.g. assuming HWE and random mating) helps with parameter identifiability/estimation problems.
If you obtain "significant results" (that you either disbelieve - because they seem "too good to be true" - or believe, in the hope that they are true!) at one or more SNPs analysed, our first recommendation is to make a smaller input data set (e.g. using the --extract snplist.txt command in PLINK) consisting just of this subset of SNPs of interest, and re-run PREMIM and EMIM just on this subset of SNPs. This will allow you to examine the output file emimresults.out more carefully (normally this file is too big to easily sort through in order to find the results pertaining to one specific SNP). You should also carefully examine the last column of the file emimsummary.out to see if there is an indicator of a warning message at any SNP. Estimated parameters with an estimated standard error (SE) of 0 in the relevant column of emimsummary.out can also suggest that there was some problem with estimating this particular parameter.
If your data was derived from a genome-wide SNP array, we recommend you follow standard GWAS QC procedures to remove unreliable samples (people) and SNPs prior to carrying out an analysis in EMIM. In addition to standard case/control QC(such as removing SNPs and individuals with large amounts of missing data, and removing SNPs with very low minor allele frequencies), we recommend you remove (or check) SNPs or families showing high rates of Mendelian misinheritance errors.
If your data was derived from a genome-wide SNP array, we also strongly recommend you check the "cluster plots" (SNP intensities) for any "significant" SNPs you obtain, in order to be sure that the genotypes have been called correctly. (This check may also be relevant for data generated using other genotyping technologies). Our experience is that poor genotype clustering (resulting in incorrect genotype calls) can produce many more apparent (but false) significant results when you apply a genotype-based test (such as modelling two child genotype effects, 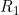 and
and 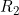 , or two maternal genotype effects,
, or two maternal genotype effects, 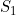 and
and 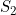 ) than when you apply an allele-based test (such as the child trend model, where is assumed to equal squared), owing to the fact that poor clustering can result in one genotype being completely or virtually absent in your data set. Although it is certainly possible that a genotype-based model may genuinely be a better model for the SNP effects in your data than an allele-based model, a discrepency between the genotype-based and allele-based results can indicate a possible problem with genotype calling.
) than when you apply an allele-based test (such as the child trend model, where is assumed to equal squared), owing to the fact that poor clustering can result in one genotype being completely or virtually absent in your data set. Although it is certainly possible that a genotype-based model may genuinely be a better model for the SNP effects in your data than an allele-based model, a discrepency between the genotype-based and allele-based results can indicate a possible problem with genotype calling.
In principal there is no reason why PREMIM and EMIM cannot be applied to imputed data (i.e. data that has been imputed on the basis of known genotypes, using a program such as MACH or IMPUTE). However, this will only work for SNPs that have been well-imputed (SNPs that are poorly imputed are likely to give rise to a large number of Mendelian errors and unreliable results). We recommend that you use standard post-imputation quality control filters to filter out low-quality SNPs/genotypes prior to performing any analysis in PREMIM and EMIM.
Particular care should be taken when analysing data that has been merged from several different studies. Note that no functionality for merging is provided within PREMIM or EMIM; any merging of data needs to be carried out prior to analysis in PREMIM/EMIM using (for example) a program such as PLINK. Special care needs to be taken when merging data for A/T or C/G SNPs that these alleles have been measured (aligned) relative to the same strand of the genome (if in doubt, it may be safer to revove such SNPs entirely). The best way to do this is to obtain assay information from the vendor who provided your genotypes. A useful list of strand alignments for commonly-used genotyping chips is provided at
http://www.well.ox.ac.uk/~wrayner/strand/
We also recommend that you aim to ensure that any merged data set consists of individuals who are well matched for ancestry and come from a single homogeneous population. (Results from separate analyses of different populations can be combined later, using meta-analysis techniques, if required).
An assumption of EMIM is that genotype data for parents of cases (or controls) is missing at random i.e. at any given SNP, there should be no systematic differences between the genotype frequencies in cases who have both parents genotyped, the frequencies in cases who have one parent genotyped and the frequencies in cases who have no parents genotyped (assuming you have enough cases within these different categories to make a comparison). Similarly for controls. By taking a careful look at the cell counts in the input data files (such as caseparenttrios.dat, casemotherduos.dat casefatherduos.dat , cases.dat etc.) you may be able to uncover problems of this sort, which could indicate genotyping discrepencies possibly combined with ascertainment effects (e.g. if your data derives from two different studies, one of which included cases with parents, and one of which included cases without parents, and the genotypes in these two studies are not really consistent/comparable).
